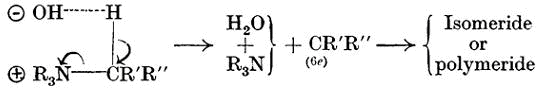
ВЛИЯНИЕ ПОЛЮСОВ И ПОЛЯРНЫХ СОПРЯЖЕНИЙ, И Т.Д. ЧАСТЬ VI. 2357
CCCVII1.–Влияние полюсов и полярных сопряжений в ряду реакций элиминации (отщепления). Часть VI. 1:1 – Элиминирование гидроокисей четвертичного аммония в процессе разложения.
КРИСТОФЕР КЕЛЬК ИНГОЛЬД и ДЖО АРТУР ДЖЕССОП
В предыдущем сообщении в части V были рассмотрены различные способы теплового разложения гидроокисей четвертичного аммония, которое сопровождается выделением олефина в среде высококипящей углеводородной смеси или выделением парафина при разложении фосфоний гидроксидов, но никогда при разложении гидроокисей аммония не наблюдается выделение других веществ, кроме олефинов. Первый из этих выводов был уже проверен; второй описывается в экспериментальной части.
Имея в виду это обстоятельство разложение в керосиновой среде зависит от анионной стабильности удаляемой группы (часть V), мы не могли придумать более подходящей структуры для благоприятного наблюдения за тепловым разложением гидроокисей четвертичного аммония чем та, что может достигнута при наличии у четвертичного атома азота 9-флуорил группы. В флуорене и его аналогах доминирующим является то обстоятельство, что отделению 9-флуоренил аниона способствует ароматическая структура (Госс и Ингольд, J., 1928, 1268. Вспомогательные факторы также способствуют этому, что иллюстрирует уникальность флуорена среди других углеводородов, так как при прямом воздействии гидроксида калия образуется соответствующяя соль.
Мы подготовили соли флуорил-9-триметил аммоня -9-триэтил и изучали разложение соответствующих гидроокисей. Ни в том, ни в другом случае не наблюдалось образование флуорена или окиси амина, на соновании чего был сделан вывод о неспособности гидроокиси аммония к тепловому разложению до парафинов.
Гидроокись флуорил-9-триметил аммония дала небольшое количество диметил-9-флуорил амина и высокий выход триметил амина; часть соответствующего количества 9-гидроксифлуорена получилась в виде 9-флуорил эфира, что соответствует наблюдениям Барбиера при синтезе этого эфира действием высокой температуры на реакционную смесь (Бер., 1575, 8, 829; Анн. Хим. Физ., 1876, 7, 507). Самое интересное, однако, состояло в том, что значительная часть материала, которая должна была выделиться в виде 9-гидроксифлуорена или соответствующего эфира, была фактически выделена в виде бис-оо/-дифениленэтилена (дифлуоренилиден), и, поскольку это не было синтезировано из гидроксида или из эфира в эквивалентных экспериментальных условиях, то это – очевидно основной продукт разложения. Гидроокись флуорил-9-триэтил аммония не дала ощутимого количества диэтил-9-флуорил амина и этилена; иначе состав вёл бы себя подобно составу триметил.
Гидроокись флуорил-9-триметил аммония дала небольшое количество диметил-9-флуорил амина и высокий выход триметил амина; часть соответствующего количества 9-гидроксифлуорена получилась в виде 9-флуорил эфира, что соответствует наблюдениям Барбиера при синтезе этого эфира действием высокой температуры на реакционную смесь (Бер., 1575, 8, 829; Анн. Хим. Физ., 1876, 7, 507). Самое интересное, однако, состояло в том, что значительная часть материала, которая должна была выделиться в виде 9-гидроксифлуорена или соответствующего эфира, была фактически выделена в виде бис-оо/-дифениленэтилена (дифлуоренилиден), и, поскольку это не было синтезировано из гидроксида или из эфира в эквивалентных экспериментальных условиях, то это – очевидно основной продукт разложения. Гидроокись флуорил-9-триэтил аммония не дала ощутимого количества диэтил-9-флуорил амина и этилена; иначе состав вёл бы себя подобно составу триметил:
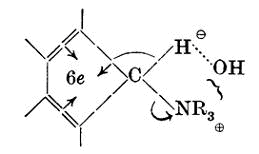
Однако, возможно представить условия, при которых группы атаковали атом α-углерода до активизации α-протона настолько эффективно, что его реакционная способность преодолевает тормозящий эффект стабильности октета, и реакция, изображённая на схеме, имеет место (продвижение, например, к R'R"C:CR'R"). Немногие группы способны к достижению этого результата, но флуореновая структура, вследствие специфического строения, упомянутого выше, очевидно способна на это.
Экспериментальная часть
9-бромфлуорен. 9-гидроксифлуорен (15 г), приготовленный из флуорена как описано Вернером и Гробом (Бер., 1904, 37, 2895), был растворён в минимальном количестве ледяной уксусной кислоты при 30–35oС, смешан с равным объемом раствора бромистого водорода в ледниковой уксусной кислоте (концентрация 600 г/л); реакционная масса была оставлена на ночь при комнатной температуре (выделение кристаллов). 9-бромфлуорен (20 г, ТПЛ=103–104.5oС), собранный после добавления к смеси равного объема воды, не требовал никакой очистки (сравните со Стаудингером, Бер, 1906, 39, 3061).
Соли флуорил-9-триэтил аммония. Бромфлуорен (6.1 г) перемешивался со спиртовым раствором триэтиламина (20 мл; концентрация 330 г/л) до тех пор пока соль не растворилась и не прекратилось выделение теплоты. Затем смесь грелась при 50oС в течение 1 часа, и бромид флуорил-9-триэтил аммония (ТПЛ=189–190oС, с разложением) был осаждён добавлением эфира, собран и промыт эфиром (найдено: Br, 24.8, 24.8. C16H18NBr вычислено Br, 26.3 %). Пикрат, полученный добавлением бромида к водному пикрата натрия, осаждался из спиртового раствора в виде пластин, которые подплавлялись при 170oС и плавились при 175oС (найдено: C, 58.0; H, 4.6. C22H20O7N4 вычислено C, 58.3; H, 4.6 %).
Разложение гидроокиси флуорил-9-триметил аммония. Раствор гидроокиси, который был приготовлен из бромида и гидроокиси серебра, имел интенсивную фиолетовую окраску*, которая исчезла вскоре после начала концентрирования; затем проводили быстрое разложение в металлическом тигле, которое завершалось при 170оС. Красно-коричневый тягучий остаток, и та часть летучих продуктов, которые не конденсировались с дистиллятом, был уловлен в ловушке с соляной кислотой; при этом никакой газ не проходил через кислотные ловушки во время процесса разложения, который сопровождался периодическим титрованием дистиллята, и по окончании реакции установка продувалась азотом. Метиловый спирт нельзя было достоверно выделить перегонкой нейтрализованного дистиллята и первичной обработкой п-нитробензоил хлоридом и пиридином. Формальдегид, который должен был образоваться из окиси триметиламина при температуре эксперимента, также не был обнаружен. Летучие продукты были извлечены (94 %) из продукта перегонки и в содержимом кислотных ловушек был в основном триметиламин (в ловушках были соляная и пикриновая кислоты), и небольшое количество примесей, включая следы основания, ограниченно растворимом в воде (возможно диметил-9-флуорил амин). Из тягучего остаток были экстрагированы кипящей водой растворимые примеси, которые были выделены при выпаривании, и затем обработаны горячей 5%-ой соляной кислотой. После кислотной обработки основания (мелкого осадка) и извлечения из эфира было получено небольшое количество (<1%) твердого амина, который был очищен дистилляцией и идентифицирован как диметил-9-флуорил амин, путём сравнения с образцом, который был приготовлен способом, описанным ниже (температура плавления кристаллов и смешанной пробы 49–50oС). Идентификация была далее проведена путём взаимодействия обоих образцов третичного амина с 2,4,6-тринитроанизолом в бензоле, с образованием пикрата флуорил-9-триметил аммония.
Смола, которая осталась после обработки водой и кислотой, была очищена в растворе бензола и разделена на три части. Одна из этих частей была использована для того чтобы показать, что добавление 1% флуорена может быть обнаружено путём возгонки, а также другими экспериментами. Вторая часть была проверена на наличие флуорена путём возгонки, но безрезультатно, и затем была обработана эквивалентным объёмом раствора пикриновой кислоты в бензоле. Избыток пикриновой кислоты отделили от пикрата с помощью сепарации из горячей массы, охлаждённый раствор был отделен от вязкого маточного раствора фильтрованием на пористом фарфоре; часть вещества была перекристаллизована дважды из бензола, при этом произошло образование красно-коричневых иголок, которые были идентифицированы как бис-оо/-дифенилилен-этилен пикрат прямым сравнением с подлинным образцом. Неочищенный остаток пикрата был обработан тёплым водным раствором аммиака и бис-oо/-дифенилилен-этилен был выделен и кристаллизован сначала из ледяной уксусной кислоты, а затем из хлороформо-спиртовой смеси. Бис-oо/-дифенилилен-этилен был идентифицирован с помощью непосредственного сравнения (температура плавления ярко-красных иголок и смешанной пробы 189–190oС), анализа (найдено: C, 94.9; H, 4.9. вычислено: C, 95.1; H, 4.9%), и переводом его в дибромид, который был приготовлен в растворе хлороформа, осаждён из лигроина, и кристаллизован из хлороформо-лигроиновой смеси (температура плавления почти бесцветных призм и смешанной пробы 236oС, с разложением). Остаток от смолистого осадка был выделен путём кристаллизации из уксусного ангидрида с образованием аморфной фракции, которая не была в дальнейшем исследована; красная прозрачная фракция, состоящая в значительной степени из бисдифенилен-этилена, который был выделен из соответствующего пикрата; желтая прозрачная фракция, с температурой плавления 165–180oС, которая состояла в значительной степени из 9-флуорилового эфира, была окрашена углеводородом и некоторой другой примесью (см. позже). Исчерпывающая очистка эфира не могла быть произведена эффективно, поэтому сырой продукт в среде уксусно-бензольной смеси был обработан бромистым водородом (реакция протекала при 40oС в течение 16 часов) для перевода эфира в 9-бромфлуорен. После этого к реакционной смеси была добавлена вода, при этом произошло отделение слоя бензола и промывка водой, затем провели осушку, и смешали со спиртовым раствором триметиламина. После выстоя в течении ночи смесь была подвергнута выпариванию досуха, а затем повторному выпариванию после добавления воды и выделению из теплой воды. Экстракт был обработан водным раствором пикрата натрия с выделением пикрата флуорил-9-триметил аммония. Нерастворимый в воде остаток был полукристаллическим, и кристаллизация из уксусной кислоты дала небольшое количество неизвестного вещества оранжевого цвета с температурой плавления 280oС (с разложением). Бисдифенилилен-этилен, необходимый для сравнения, был приготовлен по методу Грабе и Стиндта (Аннален, 1896, 291, 2).
Диметил-9-флуориламин был получен путём нагревания 9-бромфлуорена с 20% избытком водно-спиртового раствора диметиламина при 70–80oС в течение 3 часов, а затем выливанием реакционной смеси в воду. Основание, которое имело очень хорошую растворимость в безводных растворителях, и выделялось в виде масла из водно-спиртовых растворов, было наиболее легко очищено дистилляцией как указано выше (найдено: C, 86.0; H, 7.3. C15H15N, вычислено C, 86.1: H, 7.2%). Соответствующий пикрат был кристаллизован из спиртового раствора в виде листков, температура плавления которых 203–204oС (найдено: C, 57.5; H, 4.3. C21H18O7N4, вычислено C, 57.3; H, 4.1%).
Соли флуорил-9-триэтил аммония. Бромфлуорен (6.1г), триэтиламин (6мл) и спирт (15мл) нагревались вместе с обратным холодильником на паровой бане в течение 3 часов. Бромид четвертичного аммония был выделен путём добавления эфира, затем был растворён в спирте и повторно выделен из эфира. Пикрат, приготовленный из бромида и водного раствора пикрата натрия, был кристаллизован из спиртового раствора в виде пластин с температурой плавления 166oС (найдено: C, 60.5; H, 5.2. C25H26O7N4, вычислено: C, 60.7; H, 5.3 %).
Разложение гидроксида флуорил-9-триэтил аммония. Фиолетовый раствор гидроокиси концентрировался отгонкой как в предыдущем случае, с похожими результатами за исключением того, что не был получен диэтил-9-флуорил амин. Пикрат триэтиламина имел температуру плавления 175oС (Иерусалим, J., 1909, 95, 1281, описано 173oС). Бис-oo/-дифенилилен-этилен (найдено: C, 94.7; H, 5.0%) и смешанная проба имели температуру плавления 189–190оС, то есть 194–195oС с поправкой (Грабе и Мариц, Аннален, 1896, 290, 241, записано 187–188oС с поправкой).