
Золь-гель химия и технология
Кэн Мауритц
Перевод с английского: Карпенко Е.И.
Источник: Mauritz Research Group
http://www.psrc.usm.edu/mauritz/solgel.html
Процесс, который в последние годы завоевал большую известность в стекольной и керамической областях, - это золь-гель реакции. Эта технология позволяет производить разнообразные неорганические пленки из кремния или металлических алкоксильных мономерных прекурсоров. Этот метод впервые был обнаружен в конце 1800-х годов и широко изучается с начала 1930-х годов, интерес возобновился в начале 1970-х годов, когда монолитные неорганические гели были сформированы при низких температурах и превращены в стекло без высокотемпературного процесса плавления. Благодаря этому процессу могут быть получены при комнатной температуре гомогенные неорганические оксиды материалов с заданными свойствами твердости, оптической прозрачности, химической стойкости, специальной пористости, и теплового сопротивления, в отличие от намного более высоких температур плавления, необходимых в производстве обычных неорганических стекол. Определенное использование таких стекол и керамических материалов, полученных золь-гель методом, устанавливается от различных форм материала, сгенерированных в состоянии геля, т.е. монолиты, пленки, волокна и моноразмерные порошки. Много специфических назначений включают оптические, защитные и пористые пленки, оптические покрытия, периодические изоляторы, диэлектрические и электрические покрытия, высокотемпературные сверхпроводники, упрочненные стекловолокна, наполнители и катализаторы.
Золь-гель процессы, как подразумевается из названия, включают эволюцию неорганической сети через формирование коллоидной суспензии (золь) и гелеобразования золя для формирования сети в неразрывной жидкой фазе (гель). Прекурсоры для синтеза этих коллоидов состоят из металла или металлоидного элемента окруженного различными реактивными лигандами. Алкоксиды металлов являются наиболее популярными, потому что они легко реагируют с водой. Наиболее широко используются алкоксиды металлов, которые являются алкоксиланами, такие как тетраметоксилан (ТМОК) и тетраэтоксилан (ТЭОС).Однако, другие алкоксиды, такие как алюминаты, титанаты и бораты, также широко используются в золь - гель процессах, часто смешанные с ТЭОС.
На функциональном групповом уровне, как правило, используются три реакции для описания золь-гель процесса: гидролиз, конденсация алкоголя и конденсация воды. Эта общая схема реакции показана на рисунке 4. Однако, характеристики и свойства отдельных золь-гель неорганических сетей связаны с рядом факторов, которые оказывают воздействие на скорость гидролиза и реакции конденсации, это, например: рН, температура и время реакции, концентрация реагентов, природа и концентрация катализаторов , H2O/Si молярное отношение (R), температура и время выдержки и сушки. Из вышеперечисленных факторов наиболее важными являются: рН, природа и концентрация катализатора, H2O/Si молярное отношение(R) и температура. Таким образом, контролируя эти факторы, можно изменять структуру и свойства золь-гель производных неорганических сетей в широком диапазоне. Например, Сакка и др. отметили, что гидролиз ТЭОС с использованием значения R = 1-2 и М (HCl)= 0,01 в качестве катализатора дает вязкий, скручивающийся раствор. Также было показано, что эти растворы показывают высокую зависимость концентрации присущую вязкости и сильную степенную зависимость приведенной вязкости от величины среднего молекулярного веса:
[n] = k (Mn) a (l)
Значения находятся в диапазоне от 0,5 до 1,0, что указывает на линейные или немного разветвленные молекулы или цепи.
Напротив, когда значения R больше, чем 2, и / или были использованы основные катализаторы, были получены растворы, которые при эквивалентной вязкости не были скручивающимися. Значения в уравнении 1 колеблются от 0,1 до 0,5, что свидетельствует о сферической или дисковой форме частиц. Эти результаты согласуются со структурами, которые возникают при подготовке SiO2 порошков. Далее было показано, что в гидролизе при основных условиях и значениях R в диапазоне от семи до двадцати пяти, могут быть получены монодисперсные, сферические частицы.
Вообще говоря, реакции гидролиза (уравнение 2), путем добавления воды, заменяют алкоксидные группы (OR) на гидроксильные группы (OH). Последующие реакции конденсации (формулы 3 и 4) с участием силанольных групп (Si-OH) производят силоксановые связи (Si-O-Si), а также побочные продукты: воду или спирт. При большинстве условий, конденсация начинается до завершения гидролиза. Однако, условия, такие как: рН, H2O/Si молярное отношение (R), и катализатор могут вынудить завершение гидролиза перед началом конденсации. Кроме того, так как вода и алкоксиды не смешиваются, используется взаимный растворитель, такой как спирт. Наличие такого гомогенизационного агента, алкоголя, способствует гидролизу вследствие смешиваемости алкоксида и воды. Поскольку количество силоксановых связей увеличивается, отдельные молекулы соединяются и скапливаются совместно в золе. Гель формируется, когда золь частицы соединяются или взаимно переплетаются в сеть. При сушке, захваченные летучие (вода, спирт и т.д.), улетучиваются и происходит сжатие сети что приводит к дальнейшему сгущению.
Следует подчеркнуть, однако, что добавление растворителей помимо и определенные условия реакции способствуют реакциям этерификации и деполимеризации в соответствием с обратными уравнениями (2), (3) и (4).
В следующих разделах будут обсуждаться специфические факторы, которые влияют на реакции гидролиза и конденсации золь-гель процесса. Было установлено, что некоторые параметры реакции являются более важными, чем другие. В дальнейшем, мы будем прежде всего сосредоточиваться на следующих факторах: рН, природа и концентрация катализатора, и H2O/Si молярное отношение (R).
1 Гидролиз
1.1 рН
Илер делит процесс полимеризации на три области рН: <рН 2, рН 2-7, и > рН7. Однако, независимо от рН, гидролиз происходит при нуклеофильной атаке кислорода, содержащегося в воде, на атом кремния, о чем свидетельствует реакция изотопно меченой воды с ТЭОС, который был получен только непомеченными алкоголями в обоих кислотно- и щелочно-катализируемых системах.

Это можно видеть на рис.5, показывающем, как рН влияет на скорость гидролиза.
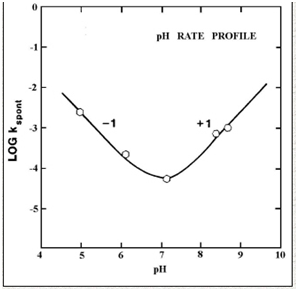
Рисунок 5 – скорость кривой рН для гидролиза в водном растворе
1.2 Тип и концентрация катализатора
Несмотря на то, что гидролиз может происходить без добавления внешних катализаторов, когда они употребляются, он протекает наиболее быстро и полно. В большинстве случаев используются минеральные кислоты (HCl) и аммиак, однако применяются и другие катализаторы: уксусная кислота, КОН, амины, KF и HF. Кроме того, было получено путем наблюдения, что на скорость и степень реакции гидролиза наибольшее влияние оказывают сила и концентрация кислотного или основного катализатора.
Аэлионом и др. было установлено, что все сильные кислоты ведут себя аналогичным образом, в то время как слабые кислоты требуют более длительного времени реакции для достижения той же степени реакции. Из графика логарифма постоянной скорости гидролиза по отношению к концентрации кислоты был получен наклон этой линии. Исследователи пришли к выводу, что реакция первого порядка в концентрации кислоты.
При основных условиях, реакции гидролиза оказалась первого порядка в концентрации основания. Однако, когда концентрация ТЭОС увеличилась, реакция отклонилась от простого первого порядка к более сложной реакции второго порядка. С более слабыми основаниями, такими как: гидроксид аммония и пиридин, измеримые скорости реакции были получены только при наличии больших концентраций. Таким образом, по сравнению с условиями кислой среды, кинетика гидролиза основания сильнее зависит от природы растворителя.
1.3 Кислотно-катализированный механизм
В кислых условиях, вполне вероятно, что алкоксидная группа является протонированной на высокоактивном первом шаге. Концентрация электронов удалена от атомов кремния, что делает его более электрофильным и, следовательно, более восприимчивы к воздействию воды. Это приводит к формированию пента-координированного переходного состояния со значительными характеристиками SN2 типа. Переходное состояние разрушается путем перемещения спирта и инверсией тетраэдра кремния, как показано на рисунке 6.

Рисунок 6 – Кислотно-катализированный гидролиз
1.4 Механизм, катализированный основанием
Катализированный основаниями гидролиз алкоголятов кремния протекает гораздо медленнее, чем кислотно-катализированный гидролиз при эквивалентной концентрации катализатора. Кислороды основных алкоксидов имеют тенденцию отталкивать нуклеофил, -OH.Однако, как только начальный гидролиз произошел, следующие реакции протекают ступенчато, причем каждая последующая алкоксидная группа легче удаляется из мономера, чем предыдущая. Вследствие этого более высокогидролизованные силиконы более склонны к атаке.Кроме того, гидролиз формирующего полимера более пространственно затруднен, чем гидролиз мономера. Хотя гидролиза в щелочной среде идет медленно, он стремится быть полным и необратимым.
Таким образом, при основных условиях, вполне вероятно, что вода диссоциирует для производства гидроксильных анионов на высокоактивном первом шаге. Гидроксильный анион затем атакует атом кремния. Опять же, был предложен механизм SN2 типа в котором -ОН вытесняет -OR с инверсией тетраэдра кремния. Это видно на рисунке 7.

Рисунок 7 – Гидролиз, катализированный основанием
1.5 H2O/Si молярное отношение (R)
Как указывалось ранее, реакция гидролиза была выполнена с R значениями в диапазоне от менее 1 до более 50, в зависимости от желаемого полисиликатного продукта. Из уравнения 2, увеличение значения R, ожидаемо содействует реакции гидролиза. Аэлионом и др. обнаружен кислотно-катализируемый гидролиз ТЭОС первого порядка в концентрации воды, однако они пронаблюдали выраженную зависимость нулевого порядка концентрации воды при основных условиях. Вероятно, это связано с производством мономеров при помощи гидролиза силоксановых связей и перераспределения реакций (например, обратные реакции 3 и 4). Однако, наиболее очевидным эффектом от увеличения значения R является ускорение реакции гидролиза. Кроме того, более высокие значения R вызывают более завершенный гидролиз мономеров, происходящий до значительных конденсаций. Различные размеры мономеров гидролиза должны влиять на относительные скорости реакции конденсаций с образованием спирта или воды. Вообще, при стехиометрических добавлениях воды (R <<2), механизм конденсационного производства спирта улучшается, в то время как реакция конденсационного формирования воды улучшается при R і 2,28.
Несмотря на то, что повышенные значения R в целом способствуют гидролизу, когда R увеличивается при сохранении постоянной растворителя: силикатного отношения, силикатная концентрация снижается. Это, в свою очередь, снижает скорости гидролиза и конденсации, приводя к увеличению времени гелирования. Этот эффект проявляется в рисунке 8, который показывает время гелирования для кислотно-катализируемых ТЭОС-систем как функцию от R и исходного алкоголя: ТЭОС - молярное отношение.
Наконец, так как вода побочный продукт реакции конденсации (уравнение, 3), большие значения R содействуют гидролизу силоксановых связей (обратимое уравнение 3).
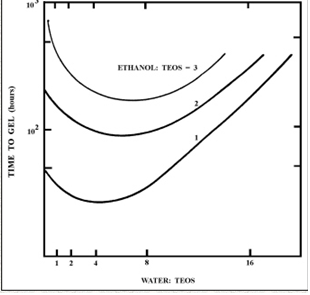
Рисунок 8 – Время гелирования как функция H2O: соотношение ТЭОС, R
2 Конденсация
2.1 рН
Полимеризации для формирования силоксановых связей происходит при помощи реакции конденсации либо с производством алкоголя, либо с производством воды. Это было показано в работе Энгельгардта и др.: типичная последовательность конденсации продуктов есть мономер, димера, линейный тример, циклический тример, циклический тетрамер, и кольца высшего порядка. Эта последовательность конденсации требует как деполимеризации (раскрытия кольца), так и наличия мономеров, которые находятся в растворе равновесно с олигомерными видами и / или образованные деполимеризацией (обратимое уравнение 3 и 4).
Скорость таких полимеризаций раскрытий кольца и реакций увеличения мономера зависит от рН среды. В полимеризации, где рН ниже 2, величина конденсации пропорциональна к [Н +] концентрации. Поскольку растворимость (см. рисунок 9) диоксида кремния является действительно низкой, ниже рН 2, образование и агрегация первичных частиц диоксида кремния происходит совместно и созревание (т. е. рост сети) мало способствует росту частиц, диаметр которых превышает 2. Таким образом, развивающиеся гелевые сети состоят из чрезвычайно малых первичных частиц.
Общим является соглашение, что между рН 2 и рН 6 величина конденсации пропорциональна [-OH] концентрации. Конденсация предпочтительно происходит между более высококонденсированными веществами и теми, которые менее конденсированы или отчасти нейтральны. Это приводит к мысли, что величина димеризации низкая, однако одиночные формы димеров реагируют преимущественно с мономерами для образования триммеров, которые, в свою очередь, реагируют с мономерами для образования тетраметров. Циклизация происходит вследствие схожести концов цепочек и значительного уменьшения распределения мономеров. Дальнейший рост происходит при добавлении материалов с более низкой молекулярной массой к более высококонденсированным материалам для формирования цепей или сетей. Растворительная способность диоксида кремния в этом диапазоне рН вновь низкая и частицы прекращают расти, когда достигают 2-4 nm в диаметре.
Выше рН 7 полимеризация происходит так же, как при рН в диапазоне между рН 2 и рН 6. Тем не менее, в этом диапазоне рН, конденсированные фрагменты ионизированы и по этой причине взаимно отталкиваются. Рост происходит главным образом посредством добавления мономеров к более высококонденсированным частицам охотнее, чем при агрегировании частиц. Вследствие большей растворимости диоксида кремния и большей зависимости размеров от растворимости при рН больше 7, частицы растут в размере и уменьшаются в количестве поскольку высокорастворимые маленькие частицы растворяются и рекристаллизируются в большие, менее растворимые частицы. Рост прекращается, когда разница в растворимости между меньшими и большими частицами становится незаметной. Этот процесс упоминается как «созревание Оствальда». Вследствие этого, размер частиц преимущественно температурно зависимый: большая температура порождает большие частицы. Кроме того, в этом рН диапазоне, скорость роста зависит от распределения размеров частиц.
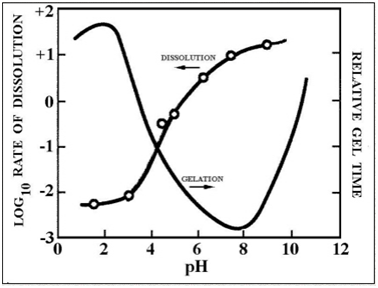
Рисунок 9 – Скорость растворения и относительное время гелирования как функция рН
2.2 Тип и концентрация катализатора
Как и в случае гидролиза, конденсация может происходить без конденсатора, однако, их использование в органосилоксанах весьма полезно. К тому же, используется такой же тип катализаторов: в большинстве случаев такие же составы, показывающие кислотные или основные свойства.
Было установлено, что реакции конденсации бывают кислотного или основного характера. В дополнение к этому, Илер установил, что при более основных условиях время гелирования зарегистрировано увеличивается. Реакции конденсации продолжают развиваться, однако, гелирование не происходит. И снова, катализатор, который диктует определенный рН, может управлять и управляет типом частицы диоксида кремния, полученной, как видно из предыдущего рассмотрения, в зависимости от рН.
2.3 Кислотно-катализированный механизм
Всеобще известно, что кислотно-катализированный механизм конденсации включает протонированные силанольные вещества. Протонация силанола приводит к большей электрофильности силикона и, таким образом, восприимчивости к нуклеофильным атакам. Более основные силанольные материалы (силанолы содержатся в мономерах или слабо разветвленных олигомерах) наиболее вероятны для протонирования. По этой причине реакции конденсации могут осуществляться преимущественно между нейтральными фрагментами и протонированными силанолами, расположенными на мономерах, конечными группами цепей, и т.д.
2.4 Механизм, катализированный основанием
Наиболее широко распространенный механизм для катализированных основанием реакций конденсации включает в себя атаки нуклеофильно депротонированного силанола на нейтральную кремниевую кислоту:

Рисунок 10 – Нуклеофильные атаки для формирования силоксановых связей
К тому же, общеизвестно, что механизм конденсации, катализированный основанием, включает пента- или гекса-координированный кремний промежуточных соединений или переходных состояний, сходных к механизму SN2 типа.
Выводы
В соответствии с Илером, золь-гель полимеризация включает в себя три стадии:
1) Полимеризация мономеров для получения частиц;
2) Рост частиц;
3) Соединение частиц в цепи, затем сети, которые удлиняются повсюду в жидкой среде, загустевая в гель.
В рамках контекста этих стадий, много факторов влияют на итоговую кремниевую сеть, такие как: рН, температура и время реакции, концентрация реагентов, тип и концентрация катализатора, H2O/Si молярное отношение (R), температура и время выдержки. Однако, в общем можно сказать, что золь-гель полученные сетки диоксида кремния, при условиях катализа кислотой, в результате являются, главным образом, линейными или случайно разветвленными полимерами, которые запутанные и формируют дополнительные ветви, приводя к гелированию. С другой стороны, сети диоксида кремния, полученные в условиях катализа основанием являются более высокоразветвленными кластерами, которые не взаимопроникают до момента гелирования, и ведут себя как дискретные кластеры.
Литература
1. http://www.psrc.usm.edu/mauritz/index2.html