Аннотация
Селективное отделение азота (N2) от метана (CH4) имеет большое значение при очистке природного газа, но добиться этого очень сложно из‐за их почти одинакового размера (диаметры молекул N2 и CH4 составляют 3,64 Å и 3,80 Å соответственно). Здесь мы теоретически изучаем адсорбцию N2 и CH4 на кластере B12 и твердых поверхностях бора α‐B12 и γ‐B28. Наши результаты показывают, что эти электронодефицитные борные материалы имеют более высокую селективность в адсорбции и захвате N2, чем CH4, что дает полезную информацию для экспериментального использования борных материалов для очистки природного газа.
Ключевые слова
Разделение N2/CH4, Очистка газа, Расчеты теории функционала плотности
1.Введение
Ожидается, что спрос на природный газ будет постоянно расти в ближайшие годы, поскольку природный газ производит меньшие выбросы CO2, чем другие виды ископаемого топлива. Новые транспортные технологии, обнаруженные значительные запасы, более низкие общие затраты и экологическая устойчивость все это указывает на то, что природный газ станет основным источником энергии в ближайшем будущем [1, 2]. Фактически, спрос на природный газ может превысить уголь к 2020 году из‐за меньшего загрязнения и более высокой эффективности использования [3]. Резервуары природного газа обычно находятся далеко от конечных рынков сбыта, и, как следствие, его приходится транспортировать либо по трубопроводам в виде газовой смеси, содержащей не менее 75% метана, либо танкерами в виде сжиженного природного газа, содержащего не менее 85% метана [4]. Выбор между двумя технологиями транспортировки зависит главным образом от расстояния и объема транспортируемого газа.
Азот является распространенным загрязнителем природного газа, и его довольно трудно удалить. Это снижает стоимость природного газа и делает его непригодным для транспортировки по большинству трубопроводов. Природный газ можно принимать для транспортировки по трубопроводам, только он содержит меньше азота, обычно от 4% до 6%. Поэтому для удаления азота было разработано несколько подходов (например, криогенное разделение, твердая адсорбция и мембранное разделение). Криогенное удаление азота является сложным и дорогостоящим процессом, не позволяющим осуществлять крупномасштабную очистку природного газа [5]. Адсорбция твердых веществ была предложена в качестве привлекательной альтернативы очистке природного газа. Однако большинство сорбентов слабо взаимодействуют с метаном и азотом и не способны эффективно их разделять [3]. Обычная мембранная технология не может эффективно отделить азот от природного газа из‐за схожих кинетических диаметров молекул метана и азота (δN2 = 3,64 Å, δCH4 = 3,80 Å)[6]. Таким образом, очень немногие материалы способны селективно адсорбировать азот из природного газа, и весьма важен поиск новых материалов с высокой селективностью и низкой стоимостью для отделения азота от природного газа.
В последние годы новые кластеры и кристаллы бора привлекли широкое внимание [7–15], благодаря своим уникальным физико‐химическим свойствам [12, 16–19]. Интерес к изучению структуры и свойств кластеров чистого бора и борсодержащих соединений растет, поскольку они имеют широкий спектр применений – от ядерных реакторов до сверхтвердых, термоэлектрических и высокоэнергетических материалов. В недавней статье Совершенство кластера бора
Граймс прокомментировал разнообразие кластеров бора, таких как нейтральные бораны, полиэдрические бораны и их производные, что побудило нас пересмотреть концепцию ковалентной химической связи [20]. Среди кластеров бора икосаэдр B12 является основной структурной единицей элементарных твердых тел бора (например, известных кристаллов α‐B12 и γ‐B28), хотя икосаэдр B12 не является устойчивым, если рассматривать его как единый изолированный кластер [21-24]. В последнее время богатые бором тройные соединения, содержащие икосаэдры B12, привлекли значительное внимание, поскольку они обладают важными особенностями как с фундаментальной, так и с практической точки зрения [7, 9, 12, 25–27].
Центральная единица кристаллического бора (т.е. икосаэдр B12) такая же, как и у многих богатых бором соединений, и может быть гибко связана, соединена или слита в жесткие каркасные структуры [12, 16–18, 21, 25, 26, 28-31]. Образование единицы B12 и ее разносторонняя связность объясняются «электронным дефицитом» или гиповалентностью бора. Для чистого элементарного бора известны только четыре кристаллические фазы: ромбоэдрическая α‐B12 [17, 26, 31] и β‐B106 [16] (с 12 и 106 атомами в элементарной ячейке соответственно), тетрагональный Т [18] (с 190–192 атомами в элементарной ячейке) и γ‐B28 (с 28 атомами в элементарной ячейке). α‐B12 состоит из одного икосаэдра B12 на элементарную ячейку, а γ‐B28 состоит из икосаэдрических кластеров B12 и пар B2 в расположении типа NaCl [12]. Кроме того, электронные свойства пар B2 и кластеров B12 в γ‐B28 различны, что приводит к переносу заряда между кластерами B12 и парами B2 [12]. В данной работе мы исследуем адсорбцию N2 и CH4 на кластере икосаэдра бора B12 и твердых поверхностях бора α‐B12 и γ‐B28. Основной мотивацией является идентификация твердых кристаллов бора в качестве новых сорбентов для очистки природного газа.
2. Вычислительные методы
Расчеты по теории функционала плотности из первых принципов [32, 33] с поправкой на дальнюю дисперсию [34] (ТФП-Д) проводились с использованием модуля DMol3 в Materials Studio [35, 36]. Кластер бора и твердые поверхности бора были полностью оптимизированы по заданной симметрии с использованием обобщенного градиентного приближения, обработанного обменно‐корреляционным потенциалом Пердью‐Берка‐Эрнцергофа. В качестве базисного набора использовалась полностью электронная двойная числовая атомная орбиталь, дополненная функциями d‐поляризации (ДПЛ). Процедура самосогласованных областей (ПСО) была использована с порога сходимости 10-6 а. е. по энергетике и электронной плотности. Прямая инверсия итерационного метода подпространства, разработанного Пулаем, была использована с размером подпространства 6 для ускорения сходимости ПСО на этих больших кластерах [37]. Для достижения сходимости ПСО, когда зазор между самой высокой занятой молекулярной орбиталью и самой низкой незанятой молекулярной орбиталью (ВЗМО – ННМО) невелик, выполняется термическое размазывание с использованием функции Ферми конечной температуры, равной 0,005 а. е. использовался. Геометрическая оптимизация была выполнена с порогом сходимости 0,002 а. е. / Å для градиента, 0,005 Å для перемещений и 10–5 а. е. для энергии. Глобальный радиус отсечения в реальном пространстве был установлен равным 4,10 Å. Для кластера B12 кластер был помещен в достаточно большую суперячейку (20 Å×20 Å×20 Å), чтобы избежать взаимодействия с его периодическими изображениями. Все параметры ячейки для α‐B12 и γ‐B28, используемые для расчетов, оптимизированы. Оптимизированные параметры ячейки для α‐B12 и γ‐B28 хорошо согласуются с экспериментальными измерениями. В деталях, оптимизированные параметры ячейки α‐B12 имеют значения α = β = γ = 5,052 Å, α = β = γ= 57,76°С, которые очень близки к значениям экспериментального измерения α = β = γ = 5,064 Å, α = β = γ = 58,10° [38]. Для γ‐B28 оптимизированными параметрами ячейки являются α = 5,042 Å, β = 5,598 Å, γ = 6,914 Å, α = β = γ = 90,0°С, которые также согласуются с экспериментальными значениями α = 5,054 Å, β = 5,612 Å, γ = 6,987 Å, α = β = γ = 90,0° [12]. Поверхности из 4×4 α‐бора (001) и 2×2 с‐бора (001) были выбраны с вакуумом 15 Å, чтобы избежать взаимодействия с его периодическими изображениями, а толщины плит α‐B12 и γ‐B28 составляют 8,012 Å и 6,914 Å соответственно. Полностью расслабленная поверхность α‐B12 (001) с клеточными векторами показана на фиг. 1. Здесь нам нужно указать, что поверхность (001) текущего исследования имеет ромбоэдрическую форму, а поверхности (001) предыдущих исследований [26, 31, 38] имеют гексагональную форму. Зона Бриллюэна была отобрана по 6×6×1 k‐точкам с использованием схемы Монкхорста‐Пака. Расчеты адсорбции N2 и CH4 на α‐B12 (001) и поверхности γ‐B28 (001) основаны на полностью оптимизированных поверхностях. Мы рассмотрели все возможные места адсорбции N2 и CH4 на поверхностях α‐B12 и γ‐B28. То, что мы обсуждали в статье, является наиболее стабильным участком адсорбции. Переходное состояние между хемосорбцией и физической адсорбцией N2 было исследовано с использованием полного ЛСП (линейного синхронного перехода) / КСП метода (квадратичного синхронного прохождения) [39], реализованного в коде Dmol3.
Энергия адсорбции N2 и CH4 на кластере B12, поверхностях α‐B12 и γ‐B28 рассчитывается по следующему уравнению:
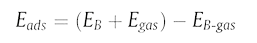
Уравнение энергии адсорбции (1)
где EB-gas – суммарная энергия адсорбента бора с адсорбированным газом, EB – энергия изолированного адсорбента бора, а Egas – энергия изолированной молекулы газа, такой как N2 и CH4. Электрон механизм распределения и передачи осуществляется по методу Малликена [40].
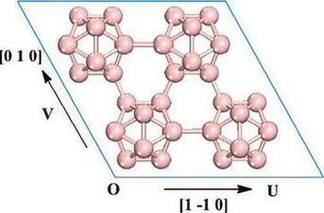
Рис. 1. Слабая поверхность α‐B12 (001) с клеточными векторами имеет ромбоэдрическую форму. Цветовой код атома: розовый, бор. (Для интерпретации ссылок на цвет в подписи к рисунку читатель может обратиться к веб ‐версии этой статьи.)
Чтобы лучше пояснить адсорбцию и природу взаимодействия N2 и CH4 на поверхностях кластеров B12, α‐B12 и γ‐B28, была использована теория атомов в молекулах (ТАМ), которая успешно использовалась для определения межмолекулярных взаимодействий различных систем, с использованием волновых функций при B3LYP/6‐311+G(d) уровне теории [41-47]. Конфигурации для расчетов ТАМ основаны на оптимизированных структурах на уровне ТФП‐Д. В анализе ТАМ, на существование взаимодействия указывает наличие так называемой критической точки связи (КТС). Прочность связи можно оценить по величине электронной плотности (ρbcp) в BCP. Аналогично, кольцевые или клеточные структуры характеризуются наличием кольцевой критической точки (КоКТ) или клеточной критической точки (КлКТ). Кроме того, природа молекулярного взаимодействия может быть предсказана по топологическим параметрам в BCP, таким как Лапласиан электронной плотности (∇2ρbcp) и плотности энергии (Hbcp). Как правило, знак ∇2ρbcp показывает, сконцентрирован ли заряд (∇2ρbcp < 0), как в ковалентных связях (совместное взаимодействие), или разряжен (∇2ρbcp > 0), как в ионных связях, H‐связях и ван‐дер‐ваальсовых взаимодействиях (взаимодействие с закрытой оболочкой). Топологический анализ системы был проведен с помощью программы AIMALL [48].
3. Результаты и обсуждение
Отделение N2 от CH4 имеет большое значение при очистке природного газа. Насколько нам известно, впервые были выполнены расчеты ТФП‐Д из первых принципов адсорбции N2 и CH4 на кластере B12, α‐B12 и γ‐B28. Наши результаты показывают, что энергии адсорбции N2 на этих материалах значительно выше, чем у CH4, что указывает на высокую селективность кристаллов бора в захвате N2 из природного газа.
3.1. Адсорбция N2 на кластере B12, α‐B12 и γ‐B28
В этой части мы обсудим результаты расчета N2 методом ТФП-Д адсорбция на икосаэдрическом кластере B12, поверхностях α‐B12 и γ‐B28. Мы начнем с адсорбции CH4 на кластере B12. Конфигурации адсорбции N2 на кластере B12 показаны на фиг. 2. Соответственно, геометрические параметры и физические и химические энергии адсорбции суммированы в таблице 1. Свободно молекула N2, длина связи N–N, по расчетам, составляет 1,109 Å. В своей физически сорбированной конфигурации (рис. 2а) N2 находится далеко от кластера B12 на расстоянии 2,990 Å. Молекулярные графики этих геометрий приведены на рис. 2а. Как показано на рис. 3а, взаимодействие – связь между кластерами N2 и B12 может быть подтверждена существованием критической точки связи (КТС) контакта N2–B. Соответствующие топологические параметры в КТС представлены в таблице 2 в качестве вспомогательной информации. Очевидно, что плотности электронов в КТС кластера N2–B между кластерами N2 и B12 невелики (Таблица 2), что указывает на то, что взаимодействие очень слабое и в основном обусловлено ван‐дер‐ваальсовыми взаимодействиями между кластерами N2 и B12. Из‐за слабого взаимодействия физически поглощенный молекула N2 (длина связи N–N = 1,110 Å) практически не подвергалась заметное структурное изменение по сравнению со свободным N2 (длина связи N–N в газовой фазе составляет 1,109 Å). Распределения зарядов по Малликену конфигураций адсорбции N2 на кластере B12 и переноса заряда между кластерами N2 и B12 перечислены в вспомогательной информации таблицы S1. Перенос заряда от N2 к кластеру B12 незначителен и составляет 0,002 э. Энергия адсорбции молекулы N2 на кластере B12, по расчетам, составляет 0,08 эВ. Кроме того, наше исследование также показывает, что процесс физической сорбции не имеет переходного состояния.
В конфигурации хемосорбции (рис. 2с) расстояние между одним атомом бора в кластере B12 и одним атомом азота в молекуле N2 должно составлять 1,515 Å. Энергия адсорбции рассчитана как 0,38 эВ на уровне PAW–PBE, что позволяет предположить, что хемосорбция является термически благоприятным процессом. При хемосорбции тройная связь молекулы N2 разорвана и слегка удлинена до 1,132 Å к верхней части B, по сравнению с молекулой N2 в газовой фазе (с длиной связи N – N 1,109 Å). Связь B – B, соединяющаяся с N2, также значительно вытягивается и удлиняется на 0,05 Å. Как только образуется хемосорбция, 0,113 отрицательного заряда самопроизвольно переходит от молекул N2 к кластеру B12 из‐за "дефицита электронов" кластера B12.
Мы выполнили расчет ЛСП/КСП, чтобы определить переходное состояние между конфигурациями физической и хемосорбции. Как показано в таблице 2, плотности электронов в BCPS для Связи N2–B физической адсорбции (рис. 2a), переходного состояния (рис. 2b) и хемосорбции (рис. 2c) постепенно увеличивались, что согласуется с процессом адсорбции от слабого взаимодействия к сильному , а расстояния между связями уменьшаются со значений 2,990 Å до 2,287 Å и 1,515 Å для трех структур соответственно. Воображаемая частота переходного состояния равна 130,4i см-1, и это присваивается режиму растяжения связи NN – B для формирования хемосорбционной конфигурации из ее физисорбционного аналога. Результаты показывают, что реагентам необходимо преодолеть барьер в 0,04 эВ на пути реакции от физической сорбции до хемосорбции. Очень низкий энергетический барьер для реакции адсорбции N2 от физической к хемосорбции указывает на то, что это кинетически благоприятный процесс.
Чтобы изучить применение кристаллов бора для разделения природного газа, мы также выполнили ТФП-Д расчеты N2 адсорбция на поверхностях α-B12 (001) и γ-B28 (001). Конфигурации адсорбции N2 на поверхностях α‐B12 и γ‐B28 показаны на рис. 2. Их важные геометрические параметры и энергии адсорбции также приведены в таблице 1. В отличие от B адсорбции N2 на кластере B12, мы получили только конфигурации хемосорбции для поверхностей α‐B12 и γ‐B28, в которых молекулы N2 плотно связаны с поверхностью α‐B12 и γ‐B28 с энергиями адсорбции 1,20 эВ и 1,07 эВ, соответственно. В своих конфигурациях тройные связи молекулы N2 разрываются, и связи N – N слегка удлиняются до 1,126 Å и 1,124 Å на верхней части B поверхностей α‐B12 и γ‐B28 соответственно. Связи B – B, связанные с N2, также значительно удлинены примерно на 0,06–0,13 Å от двух поверхностей. Расстояния между атомом B и атомом N составляют 1,469 Å и 1,479 Å для α‐B12 и γ‐B28 соответственно, что меньше, чем при адсорбции N2 на кластере B12. Это указывает на более сильные взаимодействия N2 с α‐B12 и γ‐B28, что может быть подтверждено относительно большими плотностями электронов в BCPs для связи N2–B этих двух конфигурации. Как только образуются хемосорбции, возникает 0,141 отрицательных зарядов, самопроизвольно переходящих от N2 к α‐B12 и γ‐B28 из‐за дефицита электронов
твердого бора. Наши результаты демонстрируют, что эти реакции хемосорбции не имеют переходного состояния, реакции не являются барьерными, а адсорбция кинетически благоприятна. Следовательно, адсорбция молекул N2 на поверхностях α‐B12 и γ‐B28 является энергетически и кинетически благоприятными процессами. Адсорбция N2 на поверхности α‐B12 несколько более благоприятна, чем на поверхности γ‐B28. Здесь мы должны упомянуть что исследование Макэллиготта и Робертса показало, что N2 не хемосорбируется на пленках аморфного бора [49], в то время как наши результаты расчетов показывают, что молекулы N2 могут образовывать химические связи с поверхностями кристаллов α‐B12 и γ‐B28. Причина, по которой адсорбционные свойства аморфного бора отличаются от кристаллических форм, может заключаться в том, что в аморфном боре икосаэдры бора связаны случайным образом друг с другом без порядка, и в аморфном боре будет больше деформаций и образуется больше ковалентных связей, чем в кристаллическом боре, в аморфном боре будет больше ковалентных связей. Участки адсорбции в кристаллическом боре могут иметь больше оборванных связей, чем у аморфного бора, поэтому участки адсорбции с большим количеством оборванных связей в кристаллическом боре могут образовывать сильное взаимодействие с азотом, в то время как аморфный бор не может.
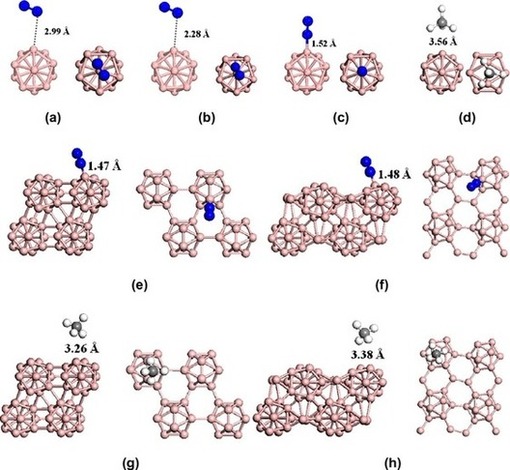
Рис. 2. (a–d) – Вид сбоку и сверху оптимизированных конфигураций адсорбции N2 и СН4 на кластере B12. (e–h) – Вид сбоку на плиты и вид сверху на поверхности оптимизированных конфигураций адсорбции N2 и СН4 на α‐B12 и γ‐B28. Цветовой код атома: синий, азот; розовый, коричневый; темно‐серый, углерод; светло‐серый, водород. (Для интерпретации ссылок на цвет в подписи к рисунку читатель может обратиться к веб‐версии этой статьи.)
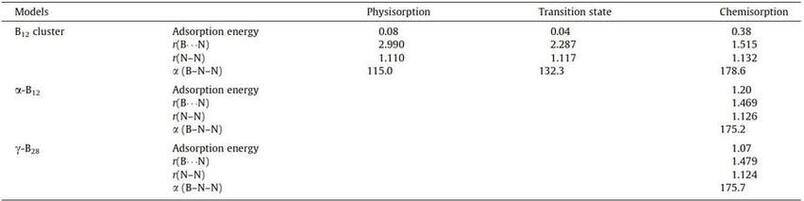
Таблица 1. Энергия адсорбции в эВ, расстояние (r) в Å и угол связи (α) в градусах для адсорбции N2 на кластере B12, поверхностях α‐B12 и γ‐B28
3.2. Адсорбция CH4 на кластерах B12, α‐B12 и γ‐B28
Чтобы понять свойства взаимодействия между материалами, содержащими бор, и молекулами СН4, мы также рассчитали адсорбцию СН4 на поверхностях кластера B12, α‐B12 и γ‐B28. Рассчитанная длина связи C – H и угол H – C – H в свободной молекуле СН4 равны 1,098 Å и 109,4° соответственно. В следующей части мы сначала обсудим адсорбцию СН4 на кластере B12. Важные структурные параметры адсорбции СН4 на кластере B12 приведены в таблице 2. Из расчета мы можем получить только СН4, адсорбированный на B12 кластеризуется по физико‐адсорбированной конфигурации. Расстояния C – B и H – B для СН4 на сорбенте составляют 3,557 Å и 2,833 Å соответственно. Мы можем видеть, что расстояние между СН4 и адсорбентом довольно большое, а энергия адсорбции составляет всего 0,08 эВ. Перенос заряда от СН4 к кластеру B12 незначителен и составляет 0,002 э (таблица S1). Эти результаты указывают на то, что их взаимодействие очень слабое и в основном возникает из‐за силы Ван‐дер‐Ваальса между СН4 и B12. Из‐за слабого взаимодействия физически сорбированный СН4 не претерпевает заметных структурных изменений по сравнению с геометрией свободного СН4. Изменения в двух связях C – H (1,098 Å) близлежащий кластер B12 незначительны по сравнению с таковыми у свободного СН4 (1,099 Å). Такая же ситуация наблюдается и для угла H – C – H, который немного уменьшается с 109,5°С до 108,9°С. Как показано на рис. 3d, взаимодействие между кластером СН4 и кластером B12 может быть подтверждено существованием критической точки связи (КТС) контакта H – B. Очевидно, что плотности электронов в КТС H – B между кластерами СН4 и B12 невелики (таблица 2). Следовательно, СН4 может быть слабо адсорбируется на кластере B12, что контрастирует с адсорбцией N2 на кластере B12.
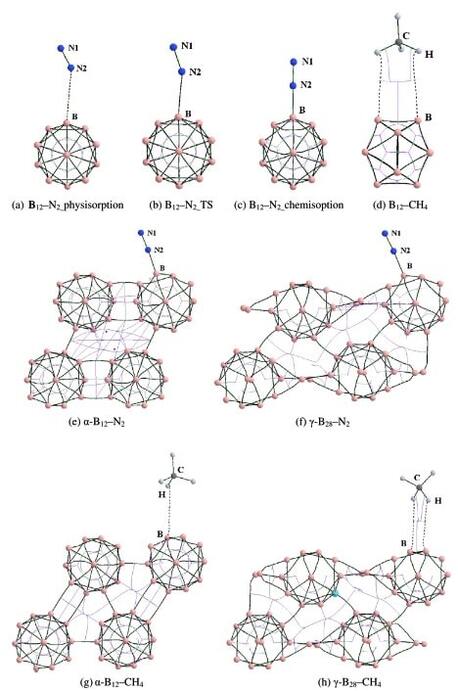
Рис. 3. Молекулярные графики промежуточных продуктов и переходного состояния адсорбции N2 и СН4 на поверхностях кластера B12, α‐B12 и γ‐B28, где критические точки связи (КТС), критические точки кольца (КТКо) и критическая точка клетки (КТКл) обозначены маленькими зелеными, красными и синие точки соответственно. (Для интерпретации ссылок на цвет в подписи к рисунку читатель может обратиться к веб‐версии этой статьи.)
Адсорбция CH4 на поверхностях α‐B12 и γ‐B28 также исследована для сравнения. Важные структурные свойства адсорбции CH4 на α‐B12 и γ‐B28 также перечислены в таблице 2. Из результатов расчетов мы видим, что расстояния между сорбентами СН4 и α‐B12, γ‐B28 довольно велики. Расстояния C – B для CH4 на α‐B12 и γ‐B28 составляют 3,255 Å и 3,380 Å соответственно, а расстояния H – B для CH4 на α‐B12 и γ‐B28 составляют 2,807 Å и 2,676 Å соответственно. Перенос заряда от СН4 к α‐B12 и γ‐B28 пренебрежимо мал и составляет 0,006 э и 0,014 э соответственно. CH4 адсорбируется на двух адсорбентах путем физической адсорбции, и энергии адсорбции на α‐B12 и γ‐B28 составляют 0,17 эВ и 0,14 эВ соответственно. Кроме того, мы можем видеть из таблицы 2, что плотности электронов на КТС связей H – B между СН4 и двумя адсорбентами малы, что согласуется с их слабыми взаимодействиями. По сравнению с взаимодействиями между N2 и двумя адсорбентами взаимодействия между CH4 и α‐B12, а также γ‐B28 очень слабы. Это демонстрирует, что α‐B12 и γ‐B28 обладают более высоким сродством к N2 и их можно использовать для отделения N2 от смеси N2/CH4.
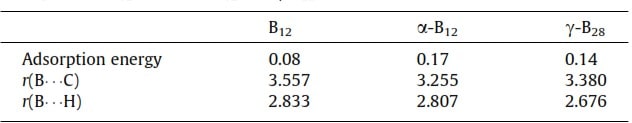
Таблица 2. Энергия адсорбции в эВ, расстояние (r) в Å и угол связи (α) в градусах для адсорбции СН4 на кластере B12, поверхностях α‐B12 и γ‐B28
Разницу в энергии адсорбции между N2 и CH4, адсорбированными на трех соединениях бора, можно понять путем анализа энергетических промежутков между их самыми высокими занятыми молекулярными орбиталями (ВЗМО) и самыми низкими незанятыми молекулярными орбиталями (ННМО). Согласно теории молекулярных орбит, пограничные орбиты и близлежащие молекулярные орбиты являются наиболее важными факторами определяющий стабильность молекулы. Чем больше разница между пограничными орбитами ВЗМО – ННМО, тем стабильнее молекулярная структура. Энергетические промежутки DE (DE = EВЗМО – EННМО) для кластера B12 поверхности α‐B12 и γ‐B28 равны 2,103 эВ, 0,046 эВ и 0,854 эВ соответственно. Ясно видно, что энергетические зазоры трех материалов на основе бора расположены в порядке кластера α‐B12 < γ‐B28 < B12. Более узкий энергетический зазор ВЗМО – ННМО означает более высокую активность молекулы. Энергетические разрывы трех борных материалов могут объяснить сила взаимодействий N2 с тремя сорбентами, которые находятся в порядке α‐B12 (энергия адсорбции 1,20 эВ) > γ‐B28 (энергия адсорбции 1,07 эВ) > кластер B12 (энергия адсорбции 0,38 эВ). Хотя энергии адсорбции СН4 на поверхностях кластера B12, α‐B12 и γ‐B28 находятся в одинаковом порядке, их значения очень малы (0,08–0,17 эВ), а взаимодействия между СН4 и всеми материалами, содержащими бор, очень слабые. Большие различия в энергиях адсорбции двух газов на двух кристаллах бора демонстрируют, что кристаллы бора являются очень хорошими материалами для разделения N2/CH4. Кроме того, селективность α‐B12 выше, чем у γ‐B28. Более того, исходя из наших результатов, мы можем предсказать, что другие твердые частицы бора с электронным дефицитом
, такие как γ‐B28 и T‐192, также могут быть использованы в качестве перспективных материалов для очистки природного газа.
4. Выводы
Таким образом, мы рассчитали адсорбцию N2 и CH4 на поверхностях кластера B12, α‐B12 и γ‐B28. Со всеми тремя материалами CH4 образует с ними слабое взаимодействие, а энергии адсорбции находятся в пределах 0,08–0,17 эВ. Однако молекулы N2 образуют с ними сильные химические взаимодействия и энергии адсорбции N2 на кластере B12, α‐B12 и γ‐B28 составляют 0,37, 1,20 и 1,07 эВ соответственно. Результаты также показывают, что адсорбция N2 на этих сорбентах бора имеет очень низкую энергию.
Список использованной литературы
- Eni sustainability report; 2006.
- Eni world oil & gas review; 2007.
- Tagliabue M, Farrusseng D, Valencia S, Aguado S, Ravon U, Rizzo C, et al. Chem Eng J 2009;155:553.
- Kidnay AJ, Parrish WR. Fundamentals of natural gas processing. Boca Raton (USA): Taylor & Francis; 2006.
- Daimiger U, Lind W. Adsorption processes for natural gas treatment: a technology update. Engelhard Brochure; 2004.
- Breck DW. Zeolite molecular sieves. New York (USA): John Willey & Sons; 1974.
- Haussermann U, Mikhaylushkin AS. Inorg Chem 2010;49:11270.
- Zhao JJ, Wang L, Li FY, Chen ZF. J Phys Chem A 2010;114:9969.
- Oganov AR, Solozhenko VL. J Superhard Mater 2009;31:285.
- Li M, Li Y, Zhou Z, Shen P, Chen Z. Nano Lett 2009;9:1944.
- Zhao YF, Lusk MT, Dillon AC, Heben MJ, Zhang SB. Nano Lett 2008;8:157.
- Oganov AR, Chen J, Gatti C, Ma Y, Ma Y, Glass CW, et al. Nature 2009;457:863.
- Bean DE, Muya JT, Fowler PW, Minh Tho N, Ceulemans A. Phys Chem Chem Phys 2011;13:20855.
- Wang Y, Robinson GH. Science 2011;333:530.
- Gonzalez Szwacki N, Sadrzadeh A, Yakobson BI. Phys Rev Lett 2007;98:166804.
- Sands DE, Hoard JL. J Am Chem Soc 1957;79:5582.
- McCarty LV, Kasper JS, Horn FH, Decker BF, Newkirk AE. J Am Chem Soc 1958;80:2592.
- Talley CP. Acta Cryst 1960;13:271.
- Szwacki NG, Sadrzadeh A, Yakobson BI. Phys Rev Lett 2007;98:166804.
- Grimes RN. J Chem Educ 2004;81:658.
- Kawai R, Weare JH. J Chem Phys 1991;95:1151.
- Boustani I. Chem Phys Lett 1995;240:135.
- Boustani I. Phys Rev B 1997;55:16426.
- Zhai HJ, Kiran B, Li J, Wang LS. Nature Mater 2003;2:827.
- He JL, Wu ED, Wang HT, Liu RP, Tian YJ. Phys Rev Lett 2005;94:015504.
- Marlid B, Larsson K, Carlsson JO. J Phys Chem B 2001;105:12797.
- Wagner P, Ewels CP, Suarez-Martinez I, Guiot V, Cox SFJ, Lord JS, et al. Phys Rev B 2011;83:024101.
- Hubert H, Devouard B, Garvie LAJ, O’Keeffe M, Buseck PR, Petuskey WT, et al. Nature 1998;391:376.
- Franz R, Werheit H. Europhys Lett 1989;9:145.
- Li D, Xu YN, Ching WY. Phys Rev B 1992;45:5895.
- Decker BF, Kasper JS. Acta Cryst 1959;12:503.
- Zhao Y, Truhlar DG. J Chem Theory Comput 2007;3:289.
- Thanthiriwatte KS, Hohenstein EG, Burns LA, Sherrill CD. J Chem Theory Comput 2011;7:88.
- Grimme SJ. Comput Chem 2006;27:1787.
- Delley BJ. Chem Phys 1990;92:508.
- Delley BJ. Chem Phys 2000;113:7756.
- Pulay PJ. Comput Chem 1982;3:556.
- Will G, Kiefer B. Z Anorg Allg Chem 2001;627:2100.
- Halgren TA, Lipscomb WN. Chem Phys Lett 1977;49:225.
- Mulliken RS. J Chem Phys 1955;23:1833.
- Perdew JP, Wang Y. Phys Rev B 1992;45:13244.
- Altarawneh M, Dlugogorski BZ, Kennedy EM, Mackie JC. J Phys Chem A 2010;114:1098.
- Altarawneh M, Kennedy EM, Dlugogorski BZ, Mackie JC. J Phys Chem A 2008;112:6960.
- Sun Q, Bu YX, Qin M. J Phys Chem A 2003;107:1584.
- Sun Q, Doerr M, Li Z, Smith SC, Thiel W. Phys Chem Chem Phys 2010;12:2450.
- Sun Q, Li Z, Du A, Chen J, Zhu Z, Smith SC. Fuel 2012;96:291.
- Sun Q, Li Z, Wang M, Du A, Smith SC. Chem Phys Lett 2012;550:41.
- Bader RFW. Atoms in molecules: a quantum theory. New York: Oxford University Press; 1990.
- McElligo Pe, Roberts RW. J Chem Phys 1967;46:273.