| Биография | Перечень ссылок | Электронная библиотека | Анализ поиска в Internet | Контакты |
| Донецкий национальный технический университет |
Попова Людмила АлександровнаАвтореферат диссертации магистра"Исследование свойств биологически активных аренсульфонатов -
Окончание магистратуры: январь 2004 года.
|
ОГЛАВЛЕНИЕ
Актуальность темы Цель работы Научная новизна Практическая ценность Введение Экспериментальная часть Обсуждение результатов Выводы Перечень условных обозначений Список ссылок |
| ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ |
| Актуальность темы |
Известно [1], что наиболее важной проблемой органической химии является взаимосвязь структуры
и реакционной способности органических молекул.
Суть данной работы заключается в исследовании неразрешенной проблемы в химии органических соединений
серы - взаимосвязи пространственных эффектов с механизмами реакций и сферами применения
пространственно затрудненных соединений. Речь идет о так называемом "положительном"
стерическом эффекте, находящем отражение в неприменимости уравнения Гаммета к реакционным
сериям с заместителями в орто-положении ароматического кольца вблизи реакционного центра.
Вследствие совмещения в одной молекуле затрудненного и сульфонильного фрагментов
получен ряд биологически активных веществ широкого спектра действия против возбудителей
капельных, гнойно-воспалительных заболеваний различной локализации, кишечных и грибковых
инфекций.
| Цель работы |
Основной целью работы является количественное изучение закономерностей щелочного гидролиза
пространственно затруденных аренсульфонатов в среде водного диоксана.
Поставленная цель
потребовала проведения следующих исследований:
| Научная новизна |
Работа является развитием теории взаимосвязи реакционной способности и структуры
органических соединений, в частности, исследования орто-эффекта алкильных групп в химии
органических соединений серы, который остается практически неисследованным явлением.
При выполнении работы была сделана попытка отойти от традиционного формального подхода
к проблеме реакционности затрудненных соединений, который не всегда может определить степень
и механизм проявления "эффекта близости". Все результаты являются новыми, приоритетными и
соответствуют мировому уровню разработок в области исследования реакционной способности
соединений и механизмов гомогенных химических реакций.
| Практическая ценность |
Большинство исследованных пространственно затрудненных серосодержащих соединений проявили
значительную антимикробную активность против возбудителей капельных, гнойно-воспалительных
заболеваний различной локализации, кишечных и грибковых заболеваний, что подтверждено
патентами (1993-1998 гг) и актами испытаний соответствующих учреждений.
Перспективные области применения: химико-фармацевтическая промышленность, медицина.
Помимо этого, исследуемые соединения представляют собой продукты химической
очистки фенолсодержащих промышленных вод методом сульфохлорирования. Образующиеся в
результате переработки фенольных стоков фенилсульфонаты после дополнительной очистки
могут использоваться в традиционных областях их применения - в качестве пластификаторов
резин и пластмасс.
Имеются данные, что рассматриваемые субстраты целесообразно применять в качестве присадок
к смазочным маслам для двигателей внутреннего сгорания.
| СОДЕРЖАНИЕ РАБОТЫ |
| Введение |
При исследовании реакций бимолекулярного нуклеофильного замещения в аренсульфохлоридах
был обнаружен эффект, определенный как "положительный стерический", заключающийся в резком
ускорении реакции гидролиза [2,3] и каталитического фенолиза [4,5] при наличии орто-метильных
заместителей вблизи сульфонильного центра. Полученные данные заставляют думать, что причина
подобного неклассического проявления стерического эффекта не ограничивается только действием
уже известных факторов заместителей и зависит не только от растворителя, а обусловлена
особенностями структуры переходного состояния SN2-типа.
| Экспериментальная часть |
Замещенные 4-нитрофениловые эфиры ароматических сульфокислот получали в щелочной среде путем обработки 4-нитрофенола или 4-нитрофенолята натрия соответствующими ароматическими сульфохлоридами. Контроль качества синтезированных веществ осуществлялся УФ-, ИК-, ПМР-спектроскопией. Растворители для кинетических измерений готовили согласно методикам [6] и использовали свежеочищенными.
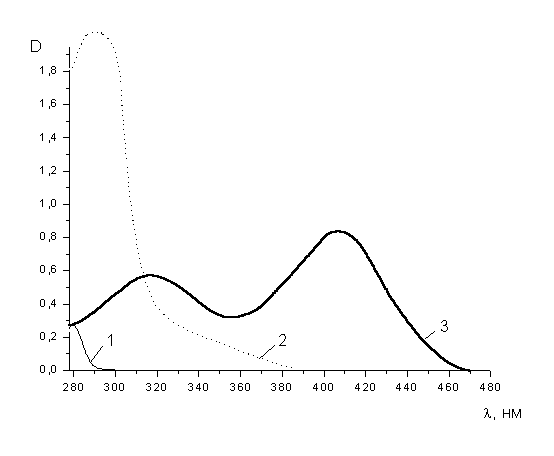
Спектры соединений в видимой и УФ-области регистрировали на спектрофотометре СФ-16
в термостатированных кварцевых кюветах толщиной 1 см в 70% водном диоксане. На рис. 1
на примере мезитиленсульфоната, соли сульфокислоты и нитрофенолята калия представлены
результаты спектроскопических измерений исходного субстрата и продуктов его гидролиза.
Выбор условий кинетического эксперимента определялся возможностью проведения сравнительного анализа с процессами замещения в сульфогалогенидах [7]. Исследовался щелочной гидролиз 4-нитрофениларенсульфонатов общей формулы XAr-SO2-O-Ph-NO2-4, где Х=2,4,6-i-Pr3, 2,6-Me2-4-t-Bu, 2,4,6-Me3, 2,6-Me2-4-i-Pr, 4-OEt, 3,4-Me2, 4-t-Bu, 2,5-Me2, 4-Me, 4-Et, 4-Сl, 4-Br, 2,4,6-Me3-3-NO2, 4-NO2 в среде 70% (объем.) водного диоксана в интервале температур 303-323 К. Реакция проводилась в условиях псевдопервого порядка по нуклеофилу. Соотношение концентраций сульфоната и щелочи в исходной смеси составляло приблизительно 1:30. Контроль за ходом реакции осуществляли спектрофотометрически при длине волны 405 нм по накоплению конечного продукта - 4-нитрофенолята калия.
Эффективные константы скорости k* (с-1) реакции рассчитывали по уравнению первого порядка:
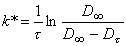
 , D
, D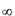 -
оптическая плотность в момент времени
-
оптическая плотность в момент времени 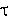 и по окончании реакции.
и по окончании реакции.
| № | X | k·102, л/(моль·с) | 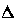 H H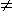 , ,кДж/моль |
-S, Дж/(моль·К) |
||
| 303 K | 313 K | 323 K | ||||
| I | 2,4,6-i-Pr3 | 0,633 | 2,17 | 6,27 | 83,0±2,4 | -13,1±7,5 |
| II | 2,6-Me2-4-t-Bu | 0,427 | 1,47 | 3,16 | 79,1±9,5 | 28,9±7,3 |
| III | 2,4,6-Me3 | 3,33 | 10,4 | 22,4 | 75,2±8,1 | 25,0±23,6 |
| IV | 2,6-Me2-4-i-Pr | 0,850 | 2,78 | 6,26 | 78,8±7,3 | 24,2±23,4 |
| V | 4-OEt | 0,895 | 2,32 | 4,94 | 66,9±0,6 | 63,3±1,2 |
| VI | 3,4-Me2 | 0,739 | 1,56 | 3,45 | 60,1±2,3 | 87,7±7,3 |
| VII | 4-t-Bu | 0,600 | 2,10 | 4,73 | 81,1±8,4 | 19,3±26,9 |
| VIII | 2,5-Me2 | 0,591 | 1,36 | 4,09 | 75,1±7,6 | 37,0±24,5 |
| IX | 4-Me | 1,59 | 3,21 | 7,33 | 59,7±4,2 | 82,6±13,3 |
| X | 4-Et | 1,20 | 2,81 | 8,32 | 76,3±6,9 | 30,6±22,0 |
| XI | 4-Сl | 6,54 | 15,0 | 27,2 | 55,4±4,3 | 84,7±13,8 |
| XII | 4-Br | 11,7 | 29,5 | 50,0 | 55,9±8,2 | 75,4±26,5 |
| XIII | 2,4,6-Me3-3-NO2 | 24,7 | 41,5 | 83,0 | 46,4±5,0 | 103±16 |
| XIV | 4-NO2 | 303 | 461 | 648 | 40,3±6,2 | 102±20 |
| Обсуждение результатов |
Обобщенную схему процесса можно представить следующим образом:
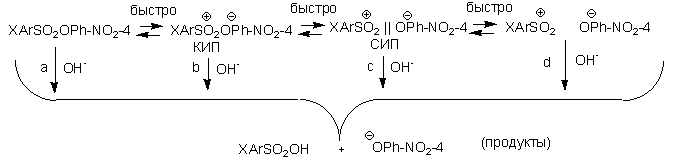 (1)
(1)Классический SN2-механизм - это реакция a. В нашем случае атака нуклеофила может осуществляться по направлениям b, c, d на различных ступенях разрыва связи S-/-O. Но все перечисленные вариации механизма затрагивают в лимитирующей стадии образование связи S…Nu, что должно проявляться в кинетических закономерностях реакции сольволиза.
Реакционная способность всех исследованных соединений повышается с возрастанием температуры
и электроноакцепторных свойств заместителя. Однако в ряде случаев накопление электронодонорных
алкильных групп в кислотной части молекулы приводит к повышению скорости реакции замещения
(I-IV, табл. 1).
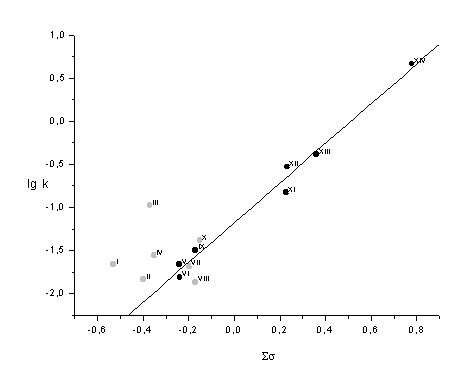
Так, для 4-нитрофенилмезитиленсульфоната (III) отношение kIII/kIX равно 3,25,
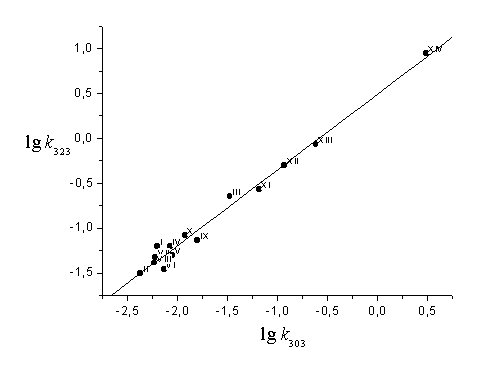
в то время как эта величина для VI и IX (табл.1) составляет 0,48. Представленная на рис. 2
изокинетическая зависимость в координатах lg kT2 - lg kT1 (Т2>Т1) [8]
и накопленный фактический материал по нуклеофильному замещению у сульфонильной серы [7, 9]
не дают оснований предполагать реализацию различных механизмов замещения в исследуемом наборе
соединений.
Ввиду разнообразия эффектов, связанных с сольватацией, электронной структурой,
пространственными требованиями исходных и переходных состояний, полученные термодинамические
параметры не дают однозначного ответа на вопрос об особенностях механизма реакции.
Тем не менее, их можно условно объединить в две группы (рис. 3).
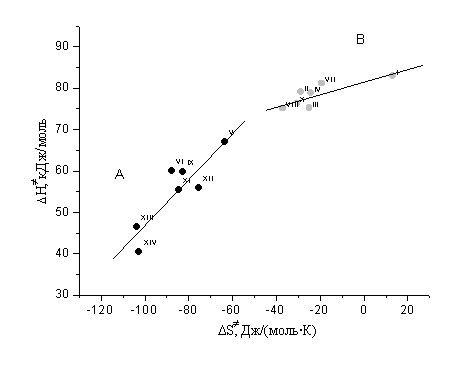 H
иS для щелочного гидролиза
аренсульфонатов XArSO2OPh-NO2-4.
H
иS для щелочного гидролиза
аренсульфонатов XArSO2OPh-NO2-4.
Первую группу (А) составляют соединения, содержащие заместители небольшого объема в положениях 3 и 4 бензольного кольца ацильной части, независимо от характера электронного влияния ( V, VI, IX, XI-XIV, табл. 1). Для них характерны изменения энтальпии и энтропии активации в пределах 40-65 кДж/моль и -(60-100) Дж/(моль·К) соответственно (рис. 3). Ко второй группе (В) относятся субстраты, имеющие объемные 2- и 4-алкильные заместители в кислотной части (I-IV, VII, VIII, X, табл.1). Энтальпия активации ПС для них изменяется в интервале 75-82 кДж/моль, энтропия активации с учетом расчетных погрешностей находится вблизи нуля (рис. 3).
Таким образом, для субстратов А наблюдается выигрыш в энтальпии активации, но переходное
состояние характеризуется определенными пространственными требованиями. Для соединений В
повышение энтальпии ПС должно приводить к понижению скорости гидролиза. Но одновременно с
этим, резкое возрастание энтропии переходного состояния свидетельствует об увеличении числа
степеней свободы ПС по сравнению с исходным состоянием. Итак, увеличение реакционной способности
системы в пределах исследуемого набора субстратов можно объяснить двумя различными факторами:
малой величиной энтальпии активации H (3- и 4-замещенные) и близкой к исходному состоянию
энтропией переходного (S~0, орто-алкильные производные).
Данный факт является достаточно
неожиданным, исходя из стерических возможностей нуклеофильной атаки атома серы для второй
группы соединений (В). Но наличие подобного аномального эффекта было обнаружено для замещения
в галогенидах аренсульфокислот [4,7].
Значение изокинетической температуры Тизо, определенное известным методом [8], оказалось
равным 433 и 150 К для первой и второй группы соединений соответственно. Не обсуждая
точность подобного определения численных значений Тизо, следует отметить следующее, являющееся
на наш взгляд основным. Рабочий интервал температур (303-323 К) находится выше или ниже
Тизо в зависимости от структурных особенностей субстратов А или В (рис. 3). Отсюда очевидно,
что интерпретация разницы реакционной способности должна быть различной при Т><Тизо.
Принято считать [10], что при Т=Тизо вклады энтальпийного H
и энтропийного T·S
факторов свободной энергии активации G уравновешиваются.
Тогда, при повышении
температуры от значения Тизо изменение H более чем
возмещается еще большим
изменением T·S, а при Т<Тизо сильные взаимодействия в
ПС снижают H ,
но одновременно существует меньше степеней свободы внутреннего движения, и, следовательно,
тем больше уменьшение S (эффект компенсации).
Можно предположить, что при Т>Тизо
(В, рис. 3) имеет место энтропийный контроль, а при Т<Тизо (А) - энтальпийный,
что отражается в кинетических закономерностях процесса гидролиза (табл. 1).
Оценка взаимосвязи реакционная способность - структура была проведена с помощью уравнения Гаммета:
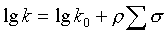
| (2) |
 - чувствительность реакционной серии к влиянию заместителей Х,
характеризуемых
- чувствительность реакционной серии к влиянию заместителей Х,
характеризуемых
 (m) или *
для пара-(мета-) и орто - заместителей соответственно.
(m) или *
для пара-(мета-) и орто - заместителей соответственно.
Субстраты А хорошо описываются этим соотношением (r=0,99, S=0,12, n=7), величина
находится в пределах 2,22-2,44, что характерно для большинства реакций бимолекулярного
нуклеофильного замещения [11]. При этом селективность системы падает при увеличении ее
реакционности (повышении температуры).
Реакционную способность субстратов второй группы (В) невозможно рассмотреть в рамках уравнения (2) (рис.3), что может быть отнесено к конкуренции влияния различных электронных и стерических факторов на скорость реакции замещения. С учетом сказанного, сделана попытка описать исследуемый ряд соединений в рамках уравнения (3):
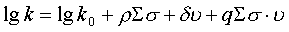
| (3) |
 - постоянная Чартона,
определяемая
из разности ван-дер-ваальсовых радиусов заместителя Х и атома водорода [8].
- постоянная Чартона,
определяемая
из разности ван-дер-ваальсовых радиусов заместителя Х и атома водорода [8].
Данные расчета приведены в табл. 2. Статистические показатели (r, S, n) уравнения
(3) являются достаточно надежными.
| T, K | -lg k0 | |
-q | r | S | n |
| 303 | 1,57±0,05 | 2,57±0,15 | 0,85±0,22 | 0,971 | 0,157 | 13 |
| 313 | 1,21±0,05 | 2,37±0,14 | 0,98±0,20 | 0,969 | 0,146 | 13 |
| 323 | 0,87±0,04 | 2,25±0,13 | 1,00±0,19 | 0,971 | 0,133 | 13 |
Коэффициент q в этом уравнении отрицателен и по значению приближается к
единице. Это означает, что дополнительно учтенный фактор
q=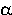 ··
··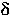 компенсирует замедление реакции для донорных заместителей, вызванное их электронными
эффектами, что может быть следствием сильной перестройки стереоэлектронной структуры
субстратов В в процессе нуклеофильной атаки. Вклад этого эффекта возрастает при
увеличении числа степеней свободы внутреннего движения в ПС (при повышении температуры)
от ~30 до 40% величины (табл. 2), что не противоречит сделанным ранее выводам,
касающимся роли фактора S для орто-замещенных субстратов.
компенсирует замедление реакции для донорных заместителей, вызванное их электронными
эффектами, что может быть следствием сильной перестройки стереоэлектронной структуры
субстратов В в процессе нуклеофильной атаки. Вклад этого эффекта возрастает при
увеличении числа степеней свободы внутреннего движения в ПС (при повышении температуры)
от ~30 до 40% величины (табл. 2), что не противоречит сделанным ранее выводам,
касающимся роли фактора S для орто-замещенных субстратов.
Обобщим сказанное. Для классического механизма (схема 1, а) характерны малые значения
энтальпии активации H . Но такой процесс требует
строгой ориентации реагентов друг относительно друга, что отражается в больших
отрицательных значениях S. Возможно,
этот механизм реализуется для мета- и пара-замещенных аренсульфонатов А (рис. 3),
поведение которых описывается уравнением (2). ПС для них можно представить следующим
образом, с учетом высокой роли образования связи S…Nu по данным уравнения (2).
Для ступенчатых процессов b, c, d (схема 1) критической ориентации атомов не требуется,
и энтропия активации положительна или приближается к нулю. В нашем случае для
субстратов В (рис. 3) разрывающаяся S-/-O связь в ПС несколько длиннее, чем в молекуле
реагента, что, вероятно, приводит к близкой к нулю величине
S. Подобное удлинение
связи S-нуклеофуг в переходном состоянии предположено ранее для сульфогалогенидов
[12] и может быть отнесено к искажению структуры субстрата при наличии орто-заместителей.
Маршрут реакции в этом случае будет приближаться к b. ПС характеризует так называемый
"промежуточный" механизм бимолекулярного замещения, где атака нуклеофила осуществляется
на сильно поляризованный субстрат.
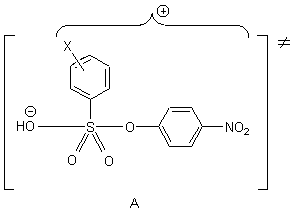
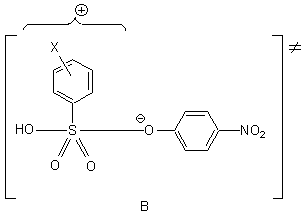
Таким образом, следует отметить, что переходное состояние при нуклеофильном замещении
в аренсульфонатах характеризуется несимметричной структурой, определяемой свойствами
заместителя. Все проявления "положительного" орто-эффекта для затрудненных сульфонильных
систем, вероятно, находятся в зоне энтропийного контроля.
| ВЫВОДЫ |
| ПЕРЕЧЕНЬ УСЛОВНЫХ ОБОЗНАЧЕНИЙ |
H- энтальпия активации S- энтропия активации - чувствительность реакционной серии к электронным эффектам заместителей - постоянная заместителя - постоянная Чартона - чувствительность реакционной серии к стерическим эффектам заместителей - мера интенсивности взаимного влияния электронных и пространственных | СПИСОК ССЫЛОК |
| <- Магистратура ДонНТУ | Главная страница ДонНТУ | Поисковая система ДонНТУ -> |