


Абсорбционные методы
Немец В.М. и др.
УДК 543.42:543.27
Н501 Спектральный анализ неорганических газов /
В.М. Немец, А.А. Петров, А.А. Соловьев. – Химия, 1988 – 240 с.: ил.
ЧАСТЬ ТРЕТЬЯ
АБСОРБЦИОННЫЕ МЕТОДЫ
ГЛАВА VI
МЕТОДЫ ПРЯМОГО ИЗМЕРЕНИЯ ПОГЛОЩЕНИЯ
Общая особенность рассматриваемых
в этой главе методов – прямое измерение ослабления интенсивности зондирующего
излучения за счет поглощения его газовой средой. Развитие этого варианта
абсорбционного спектрального анализа привело к созданию различных схем
формирования и обработки сигнала, позволяющих улучшить его аналитические
возможности:
во-первых,
путем усовершенствования способов формирования аналитического сигнала за счет
модуляции как длины волны зондирующего излучения или частоты поглощения газовых
компонентов, так и интенсивности потока излучения на приемник излучения с
помощью различных, так называемых, коррелирующих элементов;
во-вторых, путем применения
специальной обработки регистрируемого сигнала-дифференцирования его и
извлечения аналитической информации по производной от спектров поглощения.
VI.1. ОСНОВЫ МЕТОДА
VI.1.1. Общие принципы и
закономерности
Количественный абсорбционный
анализ основан на существовании зависимости между концентрацией поглощающих
атомов или молекул газа и изменением интенсивности прошедшего через
анализируемую газовую среду зондирующего излучения. Поглощение излучения
происходит на резонансных частотах, определяемых в атомах их электронными
энергетическими состояниями, а в молекулах –
электронно-колебательно-вращательными состояниями. В первом случае спектр
поглощения представляет–собой набор отдельных
спектральных линий, а во втором – набор полос, образованных совокупностью
спектральных линий.
В общем виде поглощение излучения в газе описывается законом Бугера-Ламберта
![]() (VI
1)
(VI
1)
где Iп, I0 – интенсивность поглощенного и зондирующего излучений; k(n) – спектральный коэффициент поглощения; L- толщина поглощающего слоя газа.
Поглощение газовой средой зондирующего излучения строго описывается выражением (VI 1) лишь в условиях монохроматичности излучения, независимости коэффициента поглощения от частоты и концентрации поглощающих частиц и при отсутствии фотохимических реакций в газовой среде. Вычисление концентрации поглощающих частиц возможно путем измерения величины k(n), характеризующей интенсивность линии поглощения, и параметров контура линии поглощения. Для расчета необходимо также использовать в качестве исходных предпосылок те или иные теоретические приближения, описывающие форму спектральных линий в зависимости от условий эксперимента.
В реальных условиях прямое исследование контура линии поглощения представляет весьма сложную задачу. Поэтому на практике при определении концентраций атомов и молекул измеряют интегральную интенсивность линий (полос) поглощения. Аналитический сигнал в этом случае определяется разностью интенсивностей зондирующего излучения до и после кюветы с поглощающей газовой средой. Аналитическую связь между изменением интенсивности зондирующего излучения и концентрацией поглощающих частиц находят экспериментально и используют в виде градуировочных графиков.
VI.1.2. Основные способы повышения чувствительности
Аналитические характеристики
рассматриваемого варианта абсорбционного анализа определяются прежде всего точностью регистрации и значением изменений интенсивности
прошедшего поглощающую среду зондирующего излучения и возможностью выделения
отдельных линий (полос) поглощения определяемых компонентов газовых смесей.
Решение основных проблем анализа, связанных с улучшением чувствительности и
селективности метода, достигается путем увеличения толщины поглощающего слоя
газа, повышения разрешающей способности приборов, а также использования
различных приемов формирования и обработки аналитического сигнала.
Очевидность первого способа вытекает
из выражения (VI. 1), второй способ оправдан стремлением полного выделения
аналитической линии из регистрируемого спектра поглощения. Применение этих
способов при анализе газовых сред дает хорошие результаты. Например, в работе
[187] использование многоходовой кюветы (L = 720 м) и прибора высокой
разрешающей силы (интерферометра Майкельсона) позволило определять примеси в
воздухе на уровне 10-6-10-8 % (мол.). Однако в широкой
практике только такой–прямой–путь увеличения чувствительности и селективности
не всегда возможен, да и реализация его требует применения довольно сложной
аппаратуры. Поэтому остановимся более подробно на третьем способе, включающем
различные приемы формирования и обработки аналитического сигнала.
Можно выделить по крайней мере два нетривиальных приема формирования аналитического сигнала –
дифференциальное поглощение и модуляция амплитуды сигнала. Преимущество таких
приемов заключается в изменении характера сигнала и условий измерения, а
именно: переход от регистрации малых изменений амплитуды относительно большого
постоянного сигнала к регистрации либо амплитуды сигнала на нулевом фоне, либо
меняющейся по периодическому закону амплитуды сигнала. Как известно, в этом
случае может быть достигнута значительно большая точность измерений.
Существуют также две
методики обработки сигнала: дифференцирование переменного аналитического
сигнала и расчетный метод учета мешающих наложений.
![]() (VI. 2)
(VI. 2)
где L- толщина поглощающего слоя газа
Такой метод использовался,
например, в работах [188, 189] при определении в газах примесей метана и
диоксида азота.
Второй вариант - метод двух
лучей – состоит в том, что зондирующее излучение с некоторой частотой,
желательно совпадающей с максимумом поглощения определяемого компонента,
пропускают через две идентичные кюветы, одна из которых -рабочая
– заполнена анализируемой газовой смесью, а вторая – опорная (или сравнения)
-газовой смесью известного состава. Разность сигналов опорного и рабочего
каналов есть мера концентрации определяемого компонента. Этот вариант метода
обычно используют в автоматических абсорбционных газоанализаторах, применяя электрическую или оптическую компенсации нулевого сигнала [190,
С. 156], а также в методиках, где зондирующим служит излучение диодного лазера
[191].
Модуляционный метод формирования аналитического сигнала состоит в том, что различными способами добиваются синусоидального изменения интенсивности излучения попадающего на приемник излучения. Такой модуляции можно достичь как с помощью специальных устройств, помещаемых перед приемником излучения, так и путем изменения частоты зондирующего излучения или частоты поглощения определяемых атомов или молекул.
В первом случае измеряемый сигнал зависит только от той части зондирующего излучения, которая соответствует (коррелирует) спектру поглощения определяемого компонента газовой смеси. Эта часть излучения выделяется специальными устройствами (коррелирующими элементами), пропускающими излучение только на определенных участках спектра, соответствующих структуре спектра поглощения определяемых атомов или молекул.
Такие элементы, помещенные
перед приемником излучения, обеспечивают модуляцию амплитуды регистрируемого
сигнала. В сочетании с синхронным детектированием, т. е. регистрацией сигнала в
момент, когда коррелирующий элемент выделяет только спектр поглощения
определяемого компонента, корреляционные методики позволяют существенно
ослабить влияние на результаты определения любых примесей, спектр поглощения
которых мало коррелирует по структуре с анализируемым.
В качестве коррелирующих
элементов можно использовать специальные пластинки (маски) с чередующимися
прозрачными и не прозрачными зонами, повторяющими положение линий поглощения в
плоскости изображения спектра на выходе спектрального прибора [192, 193, с.
184-202]. Модуляция амплитуды сигнала в этом случае происходит за счет
колебания маски в плоскости изображения спектра поглощения. Недостаток такой
методики модуляции сигнала – необходимость использования диспергирующей
аппаратуры с хорошим разрешением и создания целого набора масок для анализа
различных газов.
Модуляция амплитуды
зондирующего излучения может производиться также и с помощью специальных кювет
с некоторым количеством определяемого газа за счет изменения в них давления
[193, с. 184-202, 194]. В отличие от предыдущей схемы – эта более универсальна,
так как при смене аналитической задачи необходимо лишь заполнить кювету
соответствующим газом. Однако существенным ее недостатком является малая
глубина модуляции амплитуды сигнала.
По-видимому, более перспективно использование в
качестве коррелирующего элемента сканирующего интерферометра Фабри-Перо,
постоянная которого может быть выбрана в соответствии со структурой полосы
поглощения определяемого компонента газовой смеси. Длина волны максимума
пропускания интерферометра сканируется за счет изменения положения одного из
зеркал, а переход к определению нового компонента - изменением базы
интерферометра. Примером использования такого типа коррелирующих элементов
могут служить, например, работы [195, 196].
Иной принцип заложен в
методах, основанных на использовании явлений смещения частоты поглощения
молекулами или частоты излучения источников при помещении их в магнитное
(Зееман-эффект) или электрическое (Штарк-эффект) поля. В первом случае
используется явление расщепления энергетических уровней поглощающих или
излучающих атомов или молекул во внешнем магнитном поле на три (нормальный
Зееман-эффект) или большее число (аномальный Зееман-эффект) компонент. Если
источник излучения или абсорбционная кювета помещена в переменное магнитное
поле, то наблюдается соответствующее сканирование частоты зондирующего
излучения относительно линии поглощения или сканирование частоты линии
поглощения относительно частоты зондирующего излучения. В этих случаях сигнал
приемника модулируется по амплитуде с частотой изменения напряженности
магнитного поля. Как правило, в переменное магнитное поле помещают источник
излучения [197-199], реже – абсорбционную кювету [200].
Расщепление линий поглощения
в электрическом поле (Штарк-эффект) используют для определения полярных
молекул, например, аммиака [201] или диоксида серы [202]. При этом в переменное
электрическое поле помещают абсорбционную кювету с анализируемым газом.
Остановимся на специальных
способах обработки регистрируемого сигнала.
Дифференциальный метод обработки аналитического сигнала метод производной – основан на измерении первой или второй производной от меняющегося по гармоническому закону сигнала приемника. Такая методика обработки аналитического сигнала позволяет выделять слабые линии поглощения на сильном фоне и тем самым улучшать аналитические характеристики метода за счет увеличения отношения полезного сигнала к шуму. Так, в работе [203] показаны сравнительные возможности различных методик обработки регистрируемого сигнала: большие концентрации определяли методом прямого детектирования, средние – по первой, а малые до 10-7 – 10-8 % (мол.) – по второй производным. Аналогичные результаты получены и в работе [204] при определении примесей в газах на уровне 10-7 % (мол.) с регистрацией второй производной детектируемого сигнала.
Интегральный метод обработки аналитического сигнала метол учета мешающих наложений – основан на исследовании характера и интенсивности спектров поглощения анализируемых газов в некоторой области длин волн и учете их взаимных наложений. Такая методика обработки сигналов весьма трудоемка и практически невозможна без применения ЭВМ. Наиболее простой способ применен в работе [205] при анализе сложных технологических газов, где наложения учитывали путем решения системы уравнений, характеризующих вклад в поглощение на трех регистрируемых длинах волн от основных компонентов газовой смеси.
Более сложная задача
решалась в работе [206], в которой предварительные данные собирались по 225
каналам в интервале 3,8 – 5,1 мкм с шагом 0,005 мкм для бинарных смесей
определяемых газов с азотом. Затем полученные данные использовали при анализе
многокомпонентных смесей на основе азота. Еще в более общем виде решалась аналогичная задача в работе [207]. В ней рассмотрен диапазон длин
волн от 4 до 20 мкм. Исследованы наиболее интенсивные линии поглощения шести
молекул, составляющих анализируемую газовую смесь. Рассчитано интегральное поглощение
на этих линиях и вклад в него отдельных компонентов анализируемых газовых
смесей.
Рассмотренные нами методы
обработки регистрируемого сигнала, наряду с прямым детектированием изменения
интенсивности зондирующего излучения, прошедшего поглощающую газовую среду,
широко используют в различных схемах абсорбционных газоанализаторов.
VI.2. АППАРАТУРА
Важнейшие элементы
абсорбционных газоанализаторов – это источники и приемники зондирующего
излучения; их мы и рассмотрим наиболее подробно. Оптические схемы
газоанализаторов довольно просты и мы остановимся лишь на общем описании
некоторых из них.
VI.2.1. Источники зондирующего излучения
Для решения разнообразных
задач в абсорбционных газоанализаторах используют различные источники
зондирующего излучения: газоразрядные, тепловые, когерентные. По характеру
излучения их можно разделить на источники сплошного, линейчатого и
монохроматического излучения в УФ-, видимом и
ИК-спектральном диапазонах.
Тепловые источники характеризуются сплошным спектром излучения в ИК диапазоне, высокой стабильностью излучаемой мощности, малым потреблением энергии и большими сроками эксплуатации. Используют несколько разновидностей таких источников:
глобар, представляющий собой
стержень из карбида кремния; рабочая температура ~13000 К;
штифт Нернста,
представляющий собой стержень, содержащий смесь оксидов циркония, тория,
иттрия; обычная рабочая температура ~17000 К;
лампы накаливания с
вольфрамовой или нихромовой спиралью, нагретой до 10000-11000 К, излучающие в видимой и ближней ИК-областях спектра [208,
209].
Газоразрядные источники характеризуются линейчатым спектром излучения в УФ-, видимом и ближнем ИК-диапазоне длин волн, а также сплошным спектром в УФ-области спектра. К источникам этого типа относятся:
водородные [210] или дейтериевые
[195, 211] лампы, представляющие собой стеклянные колбы с кварцевыми окошками,
заполненные газом при давлении в несколько сотен Па; лампы являются источниками
сплошного спектра в видимой и УФ (до 200 нм)-областях
спектра;
высокочастотные безэлектродные
лампы, заполненные инертным газом при давлениях в несколько сотых долей Па и
веществом – источником атомных паров [212, 213]; лампы являются источниками
линейчатого спектра излучения в видимой и УФ-области;
ртутные газоразрядные лампы
низкого, высокого или сверхвысокого давления, представляющие собой кварцевые
трубки с впаянными электродами и заполненные аргоном и ртутью;
лампы являются источниками
линейчатого спектра излучения, наиболее интенсивные линии которого имеют длины волн: 253,7; 313; 314; 365,5; 404,7; 435,8; 546,1; 577 и
579,1 нм [214, 215];
лампы с
полным катодом [199, 216, 217], являющиеся источниками линейчатого спектра
излучения, характер которого определяется элементами, входящими в состав катода
или напыленного на его поверхность материала; атомы, образовавшиеся при
испарении материала нагретого катода или вследствие распыления его
поверхностных слоев под воздействием ионной бомбардировки, возбуждаются в
тлеющем разряде постоянного тока в буферном газе; эти лампы используют при
анализе воздуха на содержание металлических примесей в виде металлоорганических
соединений, аэрозолей и паров (например ртути [218]).
Монохроматические источники – оптические квантовые генераторы, излучающие отдельные линии в видимой и ИК-областях спектра в режиме импульсной или непрерывной генерации. Источники такого типа позволяют перестраивать частоту излучения либо непрерывно в некотором диапазоне длин волн, либо дискретно на нескольких фиксированных частотах:
газоразрядный He-Ne-лазер с
генерацией излучения, перестраиваемого дискретно на длинах волн 3,39; 4,22; 5,4
мкм, мощностью 0,5-5 мВт [220, 221];
лазеры на красителе (ЛК),
излучающие на длинах волн от 0,4 до 0,6 мкм [188, 196];
светодиоды на основе твердых
растворов полупроводниковых соединений типа InGaAs и InAsSbP, излучающие в
диапазоне 2,6-4,7 мкм; мощность непрерывного излучения порядка сотен мкВт, а
импульсного - нескольких мВт [222];
полупроводниковые диодные лазеры типа PbS1-x_Sex, и Pb1-xSnxSe, генерирующие в диапазоне 3-30 мкм; лазеры обеспечивают непрерывную перестройку узкой линии генерации (Dl ~ 10-6 см-1) за счет изменения тока питания и температуры полупроводникового элемента в диапазоне до 1000 см-1 [223].
В заключение упомянем о специфическом источнике излучения -СВЧ-генераторе (клистроне) с частотой 10 – 25 ГГц, используемом в некоторых газоаналитических задачах [202, 224].
Таким образом, применяемые в
абсорбционных газоанализаторах источники зондирующего излучения охватывают
широкую область спектра. Наиболее перспективно с нашей точки зрения применение
диодных полупроводниковых лазеров, позволяющих сканировать узкую линию
излучения в сравнительно большом диапазоне длин волн и генерирующих в области спектра,
перекрывающей колебательно-вращательные полосы поглощения большинства
газообразных молекул. Эти характеристики источника зондирующего излучения
обеспечивают хорошую основу для достижения высокой селективности и
универсальности газоаналитических методик.
VI.2.2. Приемники излучения
Используемые в абсорбционных
газоанализаторах приемники лучистой энергии можно разделить на две группы:
тепловые и фотоэлектрические.
Тепловые приемники служат
для детектирования излучения в ИК-области спектра (< 30 мкм). К этой группе
приемников относятся термоэлементы, представляющие собой биметаллические
устройства, при нагревании которых возникает э.д.с.,
пропорциональная температуре нагрева, а также болометры, представляющие собой
сопротивления с большим температурным коэффициентом сопротивления. Тепловые
приемники малоэффективны при измерении малых изменений мощности зондирующего
излучения и обладают относительно большой инерционностью. В качестве
положительных свойств можно указать на слабую зависимость чувствительности от
длины волны регистрируемого излучения в рабочем диапазоне и удобство в
эксплуатации.
Фотоэлектрические приемники
используют для детектирования излучения в УФ-, видимой
и ИК- (до 14 мкм) областях спектра. Эту группу приемников можно разделить на
фотоэлементы с внешним и внутренним фотоэффектом.
Первые обычно служат
приемниками излучения в УФ- и видимой областях
спектра. Принцип действия таких детекторов, называемых фотоэлектронными
умножителями (ФЭУ), основан на эмиссии с фотокатода электронов, приобретающих
от фотонов энергию, превышающую работу выхода с поверхности фотокатода.
Образовавшиеся электроны ускоряются в электрическом поле и множатся на системе
электродов — ускоряющих динодах. Сигнал ФЭУ, таким образом, пропорционален
интенсивности излучения попадающего на фотокатод приемника. Наиболее
широкое распространение получили ФЭУ с Sb-Cs-фотокатодом
с максимальной чувствительностью в области от 160 до 650 нм; с мультищелочным
фотокатодом — от 400 до 870 нм; с Ag-Cs-фотокатодом
— от 400 до 1300 нм. Постоянная времени ФЭУ составляет »10–8–10–10 с,
чувствительность »10–14 Вт.
Фотоэлектрические приемники с внутренним фотоэффектом обычно используют для регистрации излучения в ИК-области спектра. Принцип действия рассматриваемых детекторов основан на способности полупроводниковых элементов изменять свою проводимость при поглощении фотонов. В качестве таких приемников используют, например, PbS-фотосопротивления с чувствительностью в области <4 мкм или InSb с чувствительностью в области <7,5 мкм, работающие как при комнатной (293 К), так и при пониженных (273 К) температурах. В более широком спектральном диапазоне (<14мкм) могут работать детекторы типа PbSnTe или HgCdTe, но только при криогенных (»77 К) температурах.
Таким образом, применяемые в
абсорбционных газоанализаторах приемники зондирующего излучения охватывают
широкую область спектра — от 0,2 до 30 мкм. Следует отметить, что все
рассмотренные приемники являются неселективными и за исключением тепловых обладают заметной зависимостью чувствительности от
длины волны регистрируемого излучения.
VI.2.3. Схемы абсорбционных
газоанализаторов
Простейшая схема
абсорбционного газоанализатора включает:
В разд. VI.1 мы рассмотрели различные способы формирования аналитического сигнала с использованием различных коррелирующих элементов, помещаемых за абсорбционной кюветой перед приемником излучения. Кюветы с анализируемой газовой смесью имеют размеры от 1 до 500 см и часто конструируются таким образом, что обеспечивают многократное прохождение зондирующего излучения через анализируемый газ. За счет этого удается значительно увеличить толщину поглощающего слоя (например, до 720 м [187]).
Чаще всего для решения различных газоаналитических задач используют дифференциальную схему, оптическая часть которой содержит два канала с рабочей и опорной (сравнительной) кюветами. Принципиальная схема газоанализатора представлена на рис. VI.1,а. Более подробно некоторые промышленные газоанализаторы такого типа с газоразрядными источниками излучения рассматриваются в работе [190, С. 165].
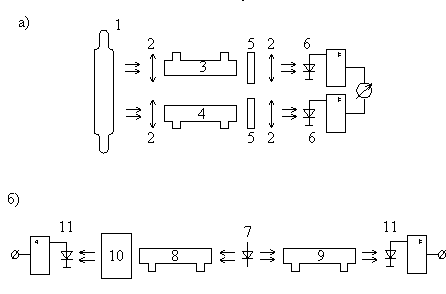
Рис. VI.1.
Схема дифференциального абсорбционного газоанализатора с газоразрядным
(тепловым) (а) и лазерным (б) источниками зондирующего излучения:
1 — газоразрядный или
тепловой источник зондирующего излучения;
2 — оптическая система
формирования пучков излучения;
3, 8 — рабочие кюветы; 4, 9
— опорные кюветы; 5— фильтры;
6 — приемники излучения; 7—
полупроводниковый лазер;
10 — диспергирующий элемент;
11 — фотоприемники
Нами кратко рассмотрены лишь основные принципиальные схемы абсорбционных газоанализаторов. В разд. VI.3 при описании различных газоаналитических методик уделяется внимание и некоторым особенностям аппаратурных схем анализаторов.
VI.3. АНАЛИТИЧЕСКИЕ
ВОЗМОЖНОСТИ МЕТОДА
Метод абсорбционного
анализа, основанный на прямом измерении поглощенной анализируемой средой
интенсивности зондирующего излучения, широко используют для решения таких
газоаналитических задач, как анализ сложных газовых смесей и изотопного состава
газовых компонентов смесей, определение примесей в чистых газах и воздухе, а
также определения металлсодержащих соединений в воздухе. Последняя задача в
настоящее время решается только с помощью рассматриваемого здесь варианта
абсорбционного метода.
VI.3.1. Анализ сложных газовых
смесей
Относительно просто решаются задачи анализа бинарных смесей. В качестве примера можно привести работы [225, 226] по определению содержания NO2 в NO в пределах от 0,01 до 5–10% (мол.) с погрешностью 20% (отн.). В обоих случаях использовалась дифференциальная схема анализатора с источником сплошного спектра излучения. В работе [225] полосы поглощения NO2 выделялись с помощью спектрофотометра типа ИКС–29, а определение NO2 в NO проводилось по поглощению на l=6,15 мкм при содержаниях от 0,01 до 1% (мол.) и на l=3,4 мкм при содержаниях от 1 до 10% (мол,). В работе [226] необходимые спектральные диапазоны выделялись с помощью фильтров.
Аналогичные схемы ИК-газоанализаторов для определения содержания СО, СО2 и СН4 в газах предложены в работах [227, 228]. Различные варианты серийного газоанализатора типа ГИАМ-5 [227] предназначены для определения в газах: СО в пределах от 5?10–3 до 100, СО2 — от 10–2 до 100 и СН4 — от 2?10–3 до 100% (мол.).
В работе [228] описывается газоанализатор, предназначенный для определения содержания СО и СО2 в потоке азота или неона в пределах от 0,5 до 10–25% (мол.). Анализаторы с источниками излучения в УФ-области спектра применяли в работе [229] для анализа газовых смесей, содержащих хлор, а в работе [230] — оксиды азота. Содержание хлора в газах определялось по поглощению излучения на l=310–350 нм в пределах от 10 до 100% (мол.) [229] с погрешностью 3–4% (отн.). В работе [230] определяемые содержания оксидов азота в смеси не превышали 1% (мол.).
Примером использования дифференциальной схемы анализатора с лазерным источником излучения в анализе сложных смесей может служить работа [222]. Диапазон излучения применявшегося лазера на основе InGaAs или InAsSbP перекрывал полосы поглощения таких молекул, как H2S (l=3,8 мкм), СН4 (l=3,32 мкм), СО2 (l=4,27 мкм), NO2 (l=4,05 мкм) и SO2 (l=3,9 мкм). В работе приводятся результаты определения содержания СН4 в газах в интервале от 10–4 до 10% (мол.) путем сравнения основного, прошедшего анализируемую смесь и совпадающего по частоте с полосой поглощения СН4, и опорного лазерных пучков.
Интересные результаты получены в работе [8] при анализе бинарных газовых смесей на основе инертных газов. Описываемые методики основаны на регистрации поглощения зондирующего излучения возбужденными атомами инертного газа, находящимися в метастабильном состоянии. Наличие в инертном газе легковозбудимых примесей приводит к ослаблению поглощения за счет уменьшения концентрации возбужденных атомов вследствие столкновений с примесными молекулами, а также изменения условий возбуждения атомов инертного газа. В работе описываются методики определения N2 в Не, Ne и Ar в пределах от 0,05 до 2% (мол.), а также Ar в Ne — от 0,1 до 1,0% (мол.). Использовалась дифференциальная схема газоанализатора, в котором источником излучения служила газоразрядная трубка с инертным газом — основой анализируемой смеси, а абсорбционная кювета, представляющая собой трубку длиной 50 см, заполнялась анализируемой смесью, в которой возбуждался разряд постоянного тока.
Из прошедшего газоразрядную абсорбционную кювету зондирующего излучения с помощью фильтров выделяли области наибольшего поглощения: l=785 (Ar), l=630 (Ne) и l=800 (Не) нм. Изменение интенсивности излучения регистрировалось с помощью ФЭУ.
Следует подчеркнуть исключительный характер описанных здесь методик, заключающийся в возможности определения как атомов инертных газов (например Ar в Ne), так и двухатомных симметричных молекул (например, N2). Решение именно таких задач газового анализа в настоящее время наиболее затруднительно методами абсорбционного анализа.
Завершая рассмотрение
возможностей метода в анализе бинарных газовых смесей, приведем некоторые
примеры использования модуляционных приемов формирования аналитического
сигнала: корреляционной, штарковской и зеемановской спектроскопии.
Примером применения первого может служить работа [196], где в качестве корреляционного элемента использовали сканирующий интерферометр Фабри-Перо. Источником излучения был ЛК с перестраиваемой генерацией в области l=450–475 нм. Частота пропускания интерферометра сканировалась за счет изменения положения одного из его зеркал, установленных на пьезокерамике. В работе приведены результаты анализа смесей, содержащих NO2 в пределах от десятых долей до одного % (мол.), и показано, что добавки других примесей (например, паров I2), имеющих сильные полосы поглощения в исследуемой области спектра, но не совпадающие по структуре с полосой поглощения молекулы NO2, не влияют на результаты определений.
Примером применения Штарк-модуляции поглощения молекул может служить работа [201], в которой описывается методика определения NH3 в воздухе на уровне 0,1–5% (мол.) с использованием в качестве источника излучения СО2-лазера (l=10,4 мкм). Анализируемая смесь прокачивалась со скоростью от 150 до 300 дм3?ч–1 через кювету длиной 20см, помещенную в электрическое поле. Поглощение модулировалось за счет синусоидального изменения напряженности электрического поля с частотой 1,2 кГц. Результаты анализов бинарных смесей с применением Зееман-модуляции поглощения определяемым компонентом смеси или частоты зондирующего излучения описаны, например, в работах [199, 200]. В работе [199] источник излучения — лампу с полым катодом, излучающую на l=213,8 нм — помещали в переменное магнитное поле с частотой 14 кГц, за счет чего длина волны источника излучения сканировалась в окрестности максимума полосы поглощения молекулы SO2. Методика позволяла определять содержание последнего в N2 на уровне не менее 1% (мол.) при длине абсорбционной кюветы 0,5 см.
В работе [200] в переменное магнитное поле помещали кювету с анализируемой смесью NO2 и N2. За счет изменения напряженности магнитного поля происходило сканирование относительно частоты зондирующего излучения максимума полосы поглощения молекул NO2. Методика позволяет определять содержание последнего в азоте от тысячных долей до десяти % (мол.).
В решении задач анализа
многокомпонентных смесей и технологических газов можно выделить, по крайней
мере, три направления. Первое — использование спектрометров с широкой
исследуемой областью спектра, позволяющих регистрировать поглощение сразу на
нескольких полосах. Это позволяет учесть возможные искажения в результатах
определений, поскольку величина искажений не может быть одинакова на различных
участках спектра.
Примером такого подхода к решению задачи анализа сложных газовых смесей могут служить работы [231, 232]. В работе [231] анализ технологических газовых смесей, содержащих оксиды азота, пары воды и азотной кислоты, проводился на ИК-спектрометре фирмы Perkin–Elmer. Газовую смесь прокачивают через кювету длиной 10 см. Оксиды азота определяли по полосам поглощения с максимумами на длинах волн: »5,24 (NO); »3,44 (NO2); »3,16 и »3,36 (N2O4) мкм. Пределы измеряемых содержаний составляли 5–15 (NO); 5–75 (NO2) и 5–35 (N2O4) % (мол.). В работе [232] для анализа смеси N2O, NO, NO2 и N2O4 использовали спектрометры типов ИКС–14А и СФ–26 с кюветами длиной 11 и 8 см. Излучение регистрировали на полосах поглощения: »0,19, »3,87, »4,03, »4,49 (N2O); »0,23, »5,22 (NO) и »0,23, »0,40, »3,42 и »6,15 (NO2); »3,18, »3,36, »3,81 и »3,87 (N2O4) мкм. Содержание компонентов смеси изменялось от сотых долей до 15% (мол.).
Второе направление заключается в выборе такого источника, длины волн монохроматического излучения которого совпадали бы с максимумами полос поглощения определяемых молекул. В работе [220] использовали излучение Не–Ne–лазера с длинами волн: 3,39, 4,22 и 5,4 мкм, совпадающих с полосами поглощения молекул СН4, СО2 и NO. Дискретное сканирование по частотам генерации проводилось за счет изменения длины резонатора лазера и введения в резонатор специального фильтра на l=3,39 мкм. Такая схема позволила определять содержание примесей этих молекул в азоте на уровне 0,1–0,5% (мол.).
В работе [233] источником излучения служила лампа с полым катодом с парами; Cd (l=214,4 нм), Zn (l=206,2 нм), Hg (l=184,9 нм) и As (l=200,3 нм). Анализировались смеси, составленные на основе N2 или Ar и содержащие примеси NO, PH3, AsH3 и SO2 на уровне от 0,1 до 10% (мол.).
Третье направление характеризуется применением различных способов учета наложений спектров посторонних молекул. Так, в работе [205] сложную технологическую смесь, содержащую NO, NO2, O2, SO2 и пары серной кислоты, анализировали путем измерения поглощения на трех длинах волн: 226 (NO); 400 (NO2) и 286 (SO2) нм. Наложения учитывались путем решения системы уравнений, оценивающих вклад в поглощение всех газовых компонентов смеси на этих длинах волн. Определяемые содержания были на уровне от 0,25 до десятков % (мол.).
В работе [206] смесь N2 с СО, СО2 и N2O анализировали на ИК-спектрометре, позволяющем регистрировать поглощения на 225 длинах волн в интервале от 3,8 до 5,1 мкм. Предварительные исследования с бинарными смесями этих газов с азотом выявили значения интенсивностей в отдельных каналах, которые в виде коэффициентов затем использовали при анализе сложных смесей. Иной вариант учета наложений использовали в работе [207] при анализе смесей, содержащих Н2О, СН4, СО, СО2, N2O и О3. В исследуемом диапазоне от 4 до 20 мкм выделены наиболее интенсивные полосы поглощения молекул 7,66 и 7,54 (СН4); 4,72 и 4,71 (СО); 7,73 (N2O); 9,55 и 9,7 (О3) мкм. Рассчитаны значения интегрального поглощения на этих длинах волн и вклад в него неопределяемых примесей, на основании чего составлена таблица учета наложений. Аналогичную схему применяли и в работе [234] при анализе сложной смеси оксидов азота (NO, N2O3 и N2O4) на содержание оксида азота. Поглощение регистрировалось в области 900–1360 нм на нескольких длинах волн: 900, 920, 950 и 1020 нм. Концентрацию NO в смеси рассчитывали с помощью эмпирического уравнения в пределах от 1 до 3% (мол.) с погрешностью 0,01% (отн.).
В заключение можно отметить,
что метод позволяет с хорошей точностью анализировать бинарные газовые смеси,
анализ же многокомпонентных смесей требует применения сложной техники учета
искажающего влияния неопределяемых компонентов.
VI.3.2. Анализ чистых газов и
воздуха
Чувствительность абсорбционного метода анализа чистых газов повышается за счет сочетания специальных способов формирования и обработки аналитического сигнала и увеличения толщины поглощающего слоя газа. В разд. VI.1.2 мы описали общие принципы таких способов, конкретные применения которых для определения микропримесей в газах рассматриваются ниже.
Пары воды Н2О определяют по поглощению излучения в ИК-области спектра (l=8,8 мкм). Методика анализа газов на содержание паров воды описана, например, в работе [203], где в качестве зондирующего использовали модулированное излучение диодного лазера (l=8,81 мкм), поглощение которого анализируемым газом происходило в многоходовой кювете, обеспечивающей эффективную толщину слоя газа до 500 м. Методика позволяет определять до 5?10–6 % (мол.) Н2О в газах. В работе [246] применяли дифференциальную схему поглощения, в которой два лазерных луча пропускали через опорную кювету с парами воды при давлении 6,7 Па и рабочую с потоком анализируемого воздуха при давлении 1,3 кПа. Газоанализатор градуировали по методу стандартных добавок в области содержаний от 2?10–7 до 10–4 % (мол.) при толщине поглощающего слоя 2 м.
Сероводород. H2S определяют по поглощению излучения в ИК-области спектра (l=7,7 мкм). Содержание H2S в воздухе на уровне 5?10–6% (мол.) определяли на спектрометре, в котором источником излучения служил штифт Нернста. Излучение на l=7,7 мкм анализируемым газом поглощалось в многоходовой кювете (L»720 м) [187].
Фторид водорода HF определяют по поглощению излучения в ИК-области спектра (l=2,35–2,85 и 24,8 мкм). В работах [248, 249], описаны методики определения микропримесей HF в газах, основанные на дифференциальных схемах поглощения: в [248] — метод двух линий, а в [249] — метод двух лучей. В первом случае [248] излучение He-Ne-лазера на l0=2,3953 мкм, совпадающей с максимумом полосы поглощения молекул HF, и на l1=2,3963 мкм, лежащей вне полосы поглощения, проходит абсорбционную кювету длиной 15 см, заполненную анализируемым воздухом при атмосферном давлении. Излучение регистрировалось фотосопротивлением типа HgCdTe, охлаждаемым до 77 К. Методика позволяет определять HF на уровне
10–5–10–6% (мол.). Другой вариант дифференциальной схемы использовали в работе [249], где излучение диодного лазера (l=2,82 или 24,8 мкм) проходит две кюветы с анализируемым и определяемым газами. В опорной кювете давление HF не превышает 0,65 кПа. Излучение, прошедшее рабочую и опорную кюветы, регистрировали охлаждаемым приемником на основе InSb. Предел определения HF по поглощению на l=2,28 мкм составляет »5?10–3, а на l=24,8 мкм — 5?10–5% (мол.).
Хлорид водорода НС1 определяют по поглощению излучения в ИК-области спектра (l=3,64 мкм). В работе [250] описывается методика определения содержания HCl в газах на уровне 10–7% (мол.) по поглощению излучения диодного лазера (l=3,64 мкм) в многоходовой кювете длиной 1,25 м (эффективная толщина слоя до 23 м) при давлении анализируемой смеси 1,3 кПа.
Метан СН4 определяют по поглощению излучения в ИК-области спектра (l=3,3–3,9 и 7,9 мкм). Примерно одинаковые результаты получены в работах [219] и [247] при использовании различных вариантов дифференциальных схем газоанализаторов: методов двух линий [219] и двух лучей [247]. В работе [219] излучение Не-Ne-лазера, генерирующего последовательность прямоугольных импульсов на l0=3,3922 и l1=3,3912 мкм, поглощалось анализируемым газом в кювете длиной 1 м. За счет стабилизации мощности излучения лазера такая схема позволила определять СН4 в газах на уровне 10–4% (мол.) с погрешностью 10% (отн.). В работе [247] излучение диодного лазера, частота генерации которого в области 7,9 мкм сканировалась за счет изменения тока питания, поглощалось в двух кюветах длиной 1 м с анализируемым и стандартным (с известной концентрацией метана) газами. Методика позволяет определять до 3?10–5% (мол.) СН4 в воздухе. Примером нелазерной методики определения СН4 может служить работа [187], в которой за счет использования многоходовой кюветы (L»700 м) получены более низкие пределы определения — 6?10–8% (мол.).
Оксид углерода СО определяют по поглощению излучения в ИК-области спектра (l=4,6–4,7 мкм). В работе [244] СО определяли в воздухе на уровне 10–4% при атмосферном давлении газа в многоходовой кювете длиной 22 м, обеспечивающей эффективную толщину поглощающего слоя до 700 м, по поглощению излучения на l=4,7 мкм при предварительной очистке воздуха от примесей Н2О и СО2. Применение более стабильного и чувствительного охлаждаемого приемника, а также ЭВМ для обработки спектров поглощения позволило при аналогичных параметрах (l=4,59 мкм, эффективная толщина слоя ~ 720 м) снизить предел определения СО в воздухе до 1?10–7% (мол.) [187].
Диоксид углерода СО2 определяют по поглощению излучения в ИК-области спектра (l=9–10 мкм). Возможности абсорбционных методик в определении микропримесей СО2 продемонстрированы в работах [203, 209]. В работе [203] источником излучения служил диодный лазер (l=9,3 мкм), что позволило определять СО2 в газах на уровне до 3?10–5% (мол.). Аналогичный результат достигнут в работе [209] [до 10–5% (мол.)] при использовании излучения теплового источника (нагретой нихромовой проволоки), но в кювете, обеспечивающей большую эффективную толщину поглощающего слоя газа — до 20 м.
Фреон-12 CF2Cl2 определяют по поглощению излучения в ИК-области спектра (l=10,8 мкм). Методика определения микропримеси фреона в воздухе описана в работе [191]. Использовали дифференциальную схему поглощения двух лучей, генерируемых диодным лазером на l=10,8 мкм в двух кюветах с анализируемым и определяемым газами. Предел определения фреона при толщине поглощающего слоя 1 м составляет 10–6–10–7% (мол.) и снижается до 10–8% (мол.) при увеличении эффективной толщины до 100 м.
Аммиак NH3 определяют по поглощению излучения в ИК-области спектра (l=8,7–9,5 мкм). Возможности определения микропримеси NH3 в газах продемонстрированы, например, в работах [203, 245]. Источником излучения в обеих работах служил диодный лазер со сканирующей частотой генерации в области 8,7–9,5 мкм. Использовали многоходовые рабочие кюветы (эффективная толщина слоя газа 500 [203] и 200 м [245]) и дополнительные кюветы сравнения для калибровки газоанализатора по определяемому компоненту. Предел определения NH3 в газах составлял 5?10–9 [203] и 10–8 [245]% (мол.).
Закись азота N2O определяют по поглощению излучения в ИК-области спектра (l=4,5 и 8,7 мкм). В работе [244] N2O в воздухе находят прямым измерением поглощения излучения на длине волны l=4,5 мкм при эффективной толщине поглощающего слоя 704 м и атмосферном давлении анализируемого газа в многоходовой кювете длиной 22 м. Методика предусматривает предварительную очистку воздуха от примесей Н2О и СО2 и позволяет определять N2O на уровне 4?10–6% (мол.). Лазерные методики определения N2O в газах описаны в работах [203, 245]. В обоих случаях длина волны генерации диодного лазера сканируется в области полосы поглощения молекулы N2O с l=8,7 мкм за счет модуляции тока питания лазера. Используемые многоходовые кюветы обеспечивают эффективную толщину поглощающего слоя газа от 200 м [245] до 500 м [203]. Методики позволяют определять примесь N2O на уровне (1–2)?10–7 % (мол.).
Оксид азота NO определяют по поглощению излучения в УФ- и ИК-областях спектра (l=214 и 226 нм и l=5,3 мкм). Практически все методики определения примесей NO основаны на применении Зееман-модулирования амплитуды сигнала как за счет сканирования в переменном магнитном поле длины волны излучения источника, так и частоты поглощения молекулы NO. Так, в работах [237, 238] излучение СО-лазера с l=5,3 мкм направляли в абсорбционную кювету, помещенную в магнитное поле, напряженность которого изменялась с частотой от 1 до 12 кГц. Давление газа в кювете не превышало 2,7–4,0 кПа. В этих условиях предел определения составил 3?10–4–5?10–5% (мол.). Иную схему использовали в работах [239–241], где в переменное магнитное поле помещали источник УФ-излучения: высокочастотную газоразрядную лампу (l=215нм) [239] или лампу с полым катодом с парами кадмия (l=214,4 и 226,5 нм) [240, 241]. В работе [239] при толщине поглощающего слоя 46 см предел определения составил 10–4% (мол.); в работах [240, 241] он был равен 1,5?10–5% (мол.) при атмосферном давлении анализируемого газа в кювете длиной 100см и 3?10–6 % (мол.) при пониженных давлениях в кювете.
Диоксид азота NO2 определяют по поглощению излучения в УФ- и ИК-областях спектра (l=249, 420–485 нм и l=6,34 мкм). В работе [188] использовали дифференциальную схему анализатора с регистрацией поглощения на двух линиях, генерируемых ЛК: l0=462,8 нм, совпадающей с максимумом полосы поглощения молекулы NO2; и l1=465,6 нм, соответствующей минимуму поглощения. Методика позволяет определять до 3?10–3% (мол.) NO2 в N2. В работе [192] использовали корреляционный метод модуляции амплитуды сигнала, а в качестве коррелирующего элемента применяли маску, нанесенную на кварцевую пластинку и повторяющую структуру полосы поглощения молекулы NO2 в области l=424,3–447,8 нм. Сигнал модулировался за счет колебания маски на выходе монохроматора в специальной вакуумной камере. Методика позволила (при толщине поглощающего слоя газа в несколько сот метров) определять примеси NO2, на уровне 10–4% (мол.). Несколько худшие результаты [10–3% (мол.)] достигнуты в работе [240], где использовали кювету длиной 100 см и источник излучения — лампу с полым катодом с парами меди (l=249,2 нм) или бора (l=249,7 и 249,8 нм), помещенную в переменное магнитное поле.
Существенное снижение предела определения [до 10–7% мол.)] достигнуто в работе [242], в которой в качестве источника излучения использовали диодный лазер с токовой модуляцией длины волны генерации в области полосы поглощения NO2 с l=6,34 мкм. Сигнал регистрировали по второй производной от спектра поглощения анализируемого газа, заполняющего многоходовую кювету длиной 1 м, что обеспечивало эффективную толщину поглощающего слоя до 40 м. Результаты работ [192] и [241] еще раз подтверждают, что при больших толщинах поглощающих слоев газа лазерные методики на несколько порядков чувствительнее методик, использующих другие источники излучения [243].
Диоксид серы SO2 определяют по поглощению излучения в УФ- и ИК-областях спектра (l=210–230 нм и l»7–9 мкм). Существующие методики определения содержания SO2 в газах используют практически все способы повышения чувствительности, при этом трудно выделить какой-либо в качестве предпочтительного. Возьмем, к примеру, результаты работ [195, 199, 202, 235], прямое сравнение которых будет наиболее корректно, так как в них используют примерно одинаковые по толщине поглощающие слои газов. В работе [195] излучение дейтериевой лампы (l»220 нм) после поглощения в абсорбционной кювете длиной 17 см направляется на коррелирующий элемент (интерферометр Фабри-Перо) и затем регистрируется ФЭУ. Предел определения газоанализатора составляет 2,5?10–3% (мол.).
Такие результаты [2?10–3% (мол.)] получены и в работе [235], где излучением диодного лазера просвечивается абсорбционная кювета длиной 10 см. Модуляция поглощения в этом случае происходит за счет сканирования частоты генерации лазера вблизи максимума полосы поглощения молекулы SO2 (l=8,8 мкм).
Следовательно, при малых толщинах поглощающих слоев возможности лазерных и нелазерных абсорбционных методик примерно одинаковы. Это подтверждается и результатами работ [199, 202], в которых использованы различные приемы модуляции аналитического сигнала, но примерно одинаковой длины поглощающие кюветы — 100см [199] и 150см [202]. Методика, где применяется источник излучения (лампа с полым катодом), помещенный в переменное магнитное поле, позволяет определять SO2 в азоте на уровне 5?10–4 [199], а с использованием Штарк-модуляции поглощения в кювете — 10–4% (мол.) [202].
Более значительные результаты достигнуты при сочетании рассмотренных выше специальных способов модуляции сигнала с увеличением эффективной толщины поглощающего слоя газа за счет применения многоходовых абсорбционных кювет. Так, в работе [204] увеличение толщины поглощающего слоя до 8,72 м и специальной обработки аналитического сигнала (регистрация по второй производной от спектра поглощения) позволило снизить предел определения SO2 в газах до 10–7% (мол.). Еще более высокий результат получен в работе [236] при использовании излучения диодного лазера мощностью 100 мкВт и многоходовой кюветы длиной 5 м. Поглощение модулировали за счет сканирования частоты генерации лазера вблизи максимума поглощения молекулы SO2 (l=7,35 мкм), что наряду с достижением значительной эффективной толщины поглощающего слоя газа (L»160 м), позволило снизить предел определения до 2?10–8 % (мол.).
Рассмотрим еще один способ повышения чувствительности метода — применение различных приемов обогащения и преобразования пробы анализируемого газа, т.е. комбинированных абсорбционных методик определения SO2 в газах. Так, в работе [210] описывается методика определения с использованием концентрирования в реакционной ячейке при пропускании анализируемого газа (проба объемом до 20 дм3) через раствор реагента. Накопленные в ячейке примеси вымывались потоком азота в абсорбционную кювету, которая просвечивалась излучением водородной лампы на l= 210 нм. Предел определения методики составляет 10–7% (мол.). Другой вариант комбинированной методики описан в работе [214], где анализируемый воздух прокачивали через реактор, содержащий ионы ртути. Продукты реакции ионов ртути с молекулами диоксида серы вымывались потоком воздуха в предварительно откалиброванный детектор паров ртути. Предел определения методики составляет 2?10–8 % (мол.).
Озон О3 определяют по поглощению излучения в ИК-области спектра (l=9,4–9,5 мкм). Возможности анализа газов на содержание озона продемонстрированы, например, в работах [187, 203]. И хотя в них использовали различные источники излучения: в работе [187] — тепловой, а в [203] — лазерный, пределы определения методик примерно одинаковы: 6?10–8 [187] и 5?10–8 [203] % (мол.). Столь низкие значения достигнуты прежде всего за счет применения многоходовых кювет, обеспечивающих значительную толщину поглощающего слоя (720 м в [187] и до 500 м [302]), а также за счет использования высокочувствительных охлаждаемых приемников.
Рассмотренные примеры довольно
полно отражают как качественный состав, так и предельные содержания примесей в
газах методом прямого измерения поглощения.
VI.3.3. Анализ изотопного
состава газовых смесей
Другой вариант дискриминации сигналов от молекул различного изотопного состава использован в работе [251]. В этом случае сигналы поглощения молекул 14NO и 15NO разделялись за счет сканирования длины волны излучения лампы с полым катодом с парами кадмия (l=214,4 и 226,5 нм) и цинка (l=213,9 нм), помещенной в переменное магнитное поле. С целью уменьшения ширины линий поглощения молекул анализ проводили при пониженных давлениях газа в кювете (Р » 16 Па). Методика позволяет определять концентрацию изотопов азота в оксиде азота в пределах от 0,37 до 98,58% (мол.).
Интересные результаты получены в работе [252] при определении изотопного состава Не. Так как резонансные линии его находятся в труднодоступной для исследований — вакуумной — области спектра, поглощение измеряли в возбужденном газе с метастабильных уровней атомов, заселяемых в газоразрядной плазме. Источником излучения служила газоразрядная трубка с анализируемым гелием. Излучение проходит абсорбционную кювету длиной 30 см, заполненную гелием естественного изотопного состава при давлении 0,27 кПа, в которой возбуждался разряд постоянного тока. Изотопный состав анализируемого гелия определялся в интервале от 1,3?10–4 до 100% (мол.) по поглощению на l=1,083 мкм.
Рассмотренные примеры
иллюстрируют, во-первых хорошие принципиальные
возможности метода прямого измерения поглощения излучения, и, во-вторых,
свидетельствуют о значительных трудностях, связанных, прежде всего, с
необходимостью исследований в области вакуумного УФ, малыми изотопическими
смещениями резонансных линий, а также со сложной структурой налагающихся
колебательно-вращательных полос поглощения изотопных молекул.
VI.3.4. Атомно-абсорбционный
анализ примесей в неорганических газах
Рассмотрим возможности
абсорбционного метода при определении в газах примесей различных металлов,
содержащихся в виде пара, аэрозольных, металлоорганических и других
соединениях. Важность решения этой задачи определяется требованиями как охраны окружающей среды, так и различных отраслей промышленности. Следует
отметить, что рассматриваемый вариант абсорбционного метода является пока
единственным аналитическим методом, позволяющим решать такого рода задачи.
Универсальный характер метода продемонстрирован, например, в работе [253], в которой описываются результаты определения микропримесей таких элементов, как Cr, Mn, Fe, Co, Ni, Cu, Zn, Cd, Pb в воздухе путем сорбции примесей на бумажных фильтрах, переводе металлов в сульфаты химическим методом, атомизации полученных растворов в пламени и регистрации поглощения на резонансных линиях. Аналогичная методика описана в работе [216] по определению содержания Са, Mg, Fe и Cu в аэрозольных и пылевых частичках в воздухе промышленных зон. Для определения использовали АА-спектрометр фирмы Перкин-Эльмер путем осаждения частиц на фильтрах, перевода их в растворы химическим методом, атомизации растворов в пламени и использовании ламп с полым катодом в качестве источников зондирующего излучения. В работе показана возможность определения содержания в воздухе:
Са l=422,7 нм 5,6×10–8–2,2×10–7% (мол.)
Mg l=285,2нм 5,5×
Fe l=248,3 нм 3,6×
Cu l=324,7 нм 1,1×
В работах [218, 254] описываются более сложные методики, включающие накопление микропримесей ртути из потока анализируемого воздуха. Так, в работе [218] пробу воздуха объемом до 6 дм3 отбирали в трубку длиной 10 см, заполненную оксидом алюминия и кварцевым песком. После отбора пробы трубку нагревали, потоком воздуха накопленная ртуть вымывалась в абсорбционную кювету длиной 10 см и по поглощению излучения ртутной лампы с полым катодом на l=253,7 нм определялось ее содержание в воздухе до 5,5?10–9% (мол.).
Более эффективный способ накопления использован в работе [254], где примесная ртуть осаждалась электростатическим способом на стенках графитовой кюветы, внутри которой помещался стальной стержень с большим отрицательным потенциалом (до 1,5 кВ) относительно стенок. Эффективность накопления при этом достигала »90%. Затем кювету нагревали и накопленную ртуть определяли по поглощению резонансной линии l=253,8 нм. Процедура определения предусматривала смешение потока анализируемого воздуха с потоком азота, обогащенного парами ртути в специальном термостате. Содержание паров ртути в смеси меняли путем изменения либо скорости потока анализируемого воздуха, либо их содержания в азоте за счет изменения температуры термостата.
Таким образом, описанная в [254] методика представляет собой вариант метода добавок, где содержание примеси в анализируемом воздухе определяется графически путем построения соответствующих зависимостей величины сигнала от содержания ртути в исследуемой газовой смеси. Методика позволяет определять содержание ртути в воздухе до 5,6?10–10% (мол.) при объеме пробы 0,2 дм3.
Различные методики определения примесных соединений Pb в воздухе описаны в работах [211, 212, 255–259]. Наиболее простая схема определения предложена в работе [255], где анализируемый воздух прокачивают через атомизатор — графитовую печь и абсорбционную кювету, в которой происходит поглощение на резонансных линиях атомами свинца излучения лампы с полым катодом. Методика позволяет определять примесь Pb на уровне до 1,7?10–9% (мол.).
В отличие от методики работы [255] большинство известных методик включает в себя различные способы накопления и разделения примесей. Так, в работах [256, 257] накопление примесей происходит на бумажных фильтрах, а атомизация — либо в ацетиленовом пламени [256], либо в графитовой печи при 2673 К [257]. В первом случае содержание Pb определяют по поглощению излучения на l=261,4 нм в пределах 10–7–10–9 % (мол.), а во втором — по поглощению излучения на l=283,3 нм на уровне до 10–10 % (мол.).
В работе [211] накопление аэрозольных частиц, содержащих соединения Pb, происходит на специальной уплотняющей поверхности, помещенной перед соплом, через которое продувается анализируемый воздух. Такая схема накопления позволяла разделять различные фракции аэрозольных частиц в зависимости от их размеров. Примеси Pb на уровне до 2?10–10 % (мол.) определяли по поглощению излучения дейтериевой лампы на l=283,3 нм. Применение более эффективного — электростатического способа накопления примесей на стенках электротермического атомизатора — позволило из объема пробы от 0,1 до 0,3 дм3 определять Pb в воздухе на уровне 3,6?10~11 % (мол.) по поглощению излучения на l=283,2 нм [258].
Комбинированные методики определения Pb в воздухе в различных соединениях путем сочетания приемов накопления, хроматографического разделения примесей, атомизации в графитовой печи и регистрации поглощения на l=283,3 нм описаны в работах [212, 259]. В работе [212] для накопления служила тефлоновая трубка длиной 30 см при 201 К, из которой затем примеси вымываются в хроматограф и после разделения поступают в электротермический атомизатор, нагретый до 1773 К. Методика позволяет определять микропримеси Pb на уровне 1,5?10–10 % (мол.). В работе [259] накопление на фильтре проводится при 143 К из потока воздуха со скоростью 6 дм3?мин–1. После хроматографического разделения примесей свинецсодержащие соединения атомизируются в графитовой печи при 2973 К в присутствии дейтерия. Методика позволяет из объема пробы до 400 дм3 определять примесь Pb на уровне 10–12 % (мол.).
В заключение приведем
описание методик определения в воздухе примесей некоторых элементов.
В работе [260] примеси селена осаждались на фильтрах из потока воздуха со скоростью до 15 дм3?мин–1. Накопленные примеси атомизировались в графитовой печи, а содержание селена находили по поглощению излучения лампы с полым катодом с парами Se на l=196 нм. Методика позволяет при объеме пробы до 45 дм3 определять примеси на уровне 3,1?10–9 % (мол.) с погрешностью от 3 до 10% (отн.) [261]. Примеси селена в воздухе определяли в виде метильных соединений [261]. Примеси разделяли на хроматографе, атомизация — в кварцевой печи при 1273 К в атмосфере смеси воздуха с водородом. Методика позволяет определять метильные соединения селена в воздухе на уровне 2,8?10–9 % (мол.).
Более эффективную схему накопления использовали в работе [263] при определении в воздухе примесей селена и теллура. Для осаждения последних служила система последовательных ловушек, первая из которых заполнялась кварцевыми позолоченными шариками. Десорбцию примесей из ловушки вели в потоке воздуха со скоростью 100 см3 -мин"1 при 1273 К. Десорбированные примеси пропускали через последующую ловушку, охлаждаемую льдом. Накопленные примеси атомизировались в кювете при 2973 К и определяли по поглощению на l = 196,0 нм для селена на уровне 2,9- 10-12 и на l = 214,3 нм для теллура – 1,7 10-12 % (мол.).
В работе [262] описывается методика определения в воздухе примесей марганца, содержащегося в аэрозольных частицах. Пробу воздуха объемом до 15м3 прокачивали через тефлоновую трубку при пониженных температурах. Накопленные примеси разделяются на хроматографе и атомизируются в кварцевой (при 1273 К) или графитовой (при 2973 К) печах. Содержание Мп определяют по поглощению излучения лампы с полым катодом с парами марганца. Методика позволяет определять примесь в воздухе на уровне 4,1?10–12 % (мол.).
Обычную для атомно-абсорбционного метода схему отбора и преобразования пробы использовали для определения примесей бериллия [264] и мышьяка [265] в воздухе: осаждение примесей на мембранном целлюлозном фильтре, перевод всего фильтра или его части в раствор, выпаривание раствора и атомизация примесных соединений в электротермическом атомизаторе при 2973 и 3073 К. При объеме пробы воздуха 90 дм3 предел определения примеси бериллия составил 1,2 10-7 (поглощение на l = 234,9 нм), а мышьяка-4,5- 10-9 % (мол.) при объеме пробы 300-400 дм3 (поглощение на l = 193,7 нм) [265].
Несколько иначе преобразуются накопленные из воздуха на целлюлозном фильтре примеси соединений олова [266]. После растворения фильтра, полученный раствор вводят в генератор гидридов, откуда гидрид олова поступает в абсорбционную кювету, нагретую до 1223 К. Поглощение зондирующего излучения измеряется на l = 224,6 нм. При объеме пробы воздуха 500 м3 предел определения методики равен 3,8-10-12 % (мол.).
В заключение можно отметить,
что в настящее время атомно-абсорбционный метод является одним из наиболее
универсальных и чувствительных для анализа неорганических газов. Недостатком же
рассмотренного варианта метода является отсутствие методик определения простых
молекул -двухатомных симметричных и одноатомных
инертных газов.
190

