
Реферат за темою випускної роботи
Зміст
- Вступ
- 1. Джерела та властивості алканів
- 2. Хімічні властивості насичених вуглеводнів
- 2.1 Реакції заміщення
- 2.2 Реакції розщеплення
- 2.3 Реакції окислення
- 2.4 Нові напрямки у вивченні реакцій алканів
- 3. Металокомплексний каталіз
- 4. Характеристика неемпіричних методів квантової хімії
- Висновки
- Перелік посилань
Вступ
Обмеженість запасів нафти і постійне подорожчання її видобутку висунули проблему пошуку альтернативної сировини для хімічної і нафтохімічної промисловості. Такою сировиною в найближчому майбутньому може стати природний газ, в першу чергу його основний компонент - метан. Метан також є побічним продуктом в цілому ряді процесів переробки органічної сировини в тому числі кам'яного вугілля, які в перспективі можуть стати головними джерелами отримання органічних напівпродуктів і моторних палив. Це висуває завдання створення ефективних процесів безпосередньої переробки метану в цінні продукти - метанол, формальдегід, нижчі олефіни і т. д.
Поряд з великою практичною важливістю, проблема переробки метану становить значний науковий інтерес, тому що пов'язана з пошуками можливості активації досить стійкою молекули, реакційна здатність якої істотно нижче, ніж цільових продуктів. [1 ]
Відносно низька реакційна здатність насичених вуглеводнів обумовлена відсутністю в молекулах цих сполук π- або n-електронів і малою полярністю σ-зв'язків С-H і С-С. Разом з тим, оскільки парафінові вуглеводні (запаси яких, особливо метану, в природі ще великі) є одним з видів хімічної органічної сировини, дуже актуальною є проблема їх переробки в функціональні похідні (спирти, кетони, кислоти), а також отримання з алканов олефінів і ароматичних вуглеводнів.
Велике практичне значення мають також процеси гідрокрекінгу й ізомеризації парафінів, що дозволяють отримувати цінні види моторного палива. Основні промислові процеси, де в якості сировини використовуються алкани, перебігають при високих температурах (вище 150-200 ° С), часто в присутності гетерогенних каталізаторів (наприклад, дегідрогенізація, ароматизація, реакція метану з водою, дає суміш СО+Н2, крекінг метану до ацетилену і водню, крекінг вищих алканів з утворенням олефінів, нітрування, фторування дією CoF3 і т. д.). Особливу область утворюють радикально-ланцюгові процеси (фотохімічне хлорування під дією Сl2 , що ініціюється світлом або радикалами окислення молекулярним киснем, сульфохлорування тощо), проводяться при низьких температурах. Однак селективність цих реакцій невелика, і зазвичай утворюються складні суміші продуктів. Нарешті, давно відома здатність деяких мікроорганізмів і клітин вищих тварин окислювати алкани киснем при звичайній температурі. Біологічне окислення зазвичай селективно. Наприклад, деякі мікроорганізми окислюють тільки кінцеву метильную групу вуглеводню, перетворюючи його первинно в спирт (ω-гідроксилювання). [2]
Дослідження процесу гетерогенно-каталітичного окиснення метану в корисні продукти можна умовно розділити на два напрями. Одне з них пов'язано з отриманням цінних кисневмісних сполук - метанолу та формальдегіду. Цей напрямок розвивається вже досить давно і тут досягнуті певні результати в розумінні механізму процесу, впливу природи активних центрів каталізаторів на їх активність і селективність. Однак виходи корисних продуктів на стабільно працюючих каталізатора не перевищують 3% по метанолу і 5-8% по формальдегіду при окисленні метану киснем або повітрям і без добавок різних сполук (Оксиди азоту, галогенопохідні) як окисників і ініціаторів. Таким чином, при гетерогенно-каталітичному окисленні метану поки не вдалося досягти суттєвого збільшення виходів цінних кисневмісних сполук в порівнянні з гомогенним окисленням.
Другий напрямок - каталітично-окисна конденсація метану у вищі вуглеводні (в основному в етан, етилен, пропілен і ацетилен) - почало розвиватися порівняно нещодавно. [1]
1. Джерела і властивості алканів
Основне джерело алканів в природі - нафта; фракції нафти 200-430 ° С містять 30-50% (за масою) насичених вуглеводнів (З них до 60% вуглеводнів нормальної будови); нижчі газоподібні насичені вуглеводні входять до складу природного газу (До 98% метану) і розчинені в нафті; тверді зустрічаються у вигляді покладів озокериту, а також утворюють воскові покриття листя, квітів і насіння рослин, входять до складу бджолиного воску.
Довжини зв'язків С-С в алканах ~ 0,154 нм, С-Н ~ 0,109 нм, кут ССС в газоподібному стані – 109,47 °, в кристалічному – на 2-3 ° більше.
Нижчі насичені вуглеводні до бутану і неопентану - гази без кольору і запаху, вуглеводні С5-С17 – безбарвні рідини з
характерним бензиновим
запахом, вищі насичені вуглеводні - безбарвні тверді речовини. Температури плавління і кипіння
залежать від розміру молекули і зростають в гомологічному ряду із збільшенням молекулярної маси. Серед ізомірів вуглеводні
нормальної будови мають найбільш високі температури кипіння і щільність. [3]
Найпростішим представником алканів є метан. Метан - безбарвний газ без запаху. Його хімічна формула – CH4. Малорозчинний у воді, легше повітря. При використанні в побуті, промисловості у метан зазвичай додають одоранти зі специфічним «запахом газу». Основний компонент природних (77-99%), попутних нафтових (31-90%), рудничного та болотного газів (звідси інші назви метану - болотний або рудничний газ). [4]
2. Хімічні властивості насичених вуглеводнів
Алкани - хімічно найменш активні органічні сполуки. Всі зв'язки С-С і С-Н в алканах одинарні, тому алкани нездатні до реакцій приєднання. Для алканів характерні реакції заміщення атомів водню на інші атоми і групи атомів.
Основні хімічні перетворення алканів йдуть тільки при повідомленні їм достатньо високої енергії (при нагріванні або опроміненні УФ-світлом). При цьому може відбутися або розрив зв'язку С-Н з подальшим заміщенням атома водню на інший атом або групу атомів.
Оскільки алкани - сполуки неполярні, то при розриві зв'язків утворюються головним чином не іони, а радикали, тобто цей процес йде за гомолітичним механізмом.
Таким чином, для алканів розрізняють два основних типи хімічних реакцій: реакції заміщення водню (з розривом зв'язку С-Н) і реакції розщеплення (з розривом зв'язків С-С і С-Н).
2.1 Реакції заміщення
Галогенування (заміщення галогеном) - найважливіша реакція алканів. Вона протікає при освітленні УФ-світлом або в темряві при сильному нагріванні, а також в присутності каталізаторів. Порівняно легко алкани вступають в реакцію заміщення з хлором і бромом, дуже важко – з йодом. З фтором реакція протікає з вибухом (тому зазвичай фтор розбавляють азотом або використовують розчинники). В результаті заміщення водню галогеном утворюються галогено-похідні алканів. Наприклад, хлорування метану перебігає з послідовним заміщенням в його молекулі всіх атомів водню на хлор.
Нітрування (заміщення нітрогрупи NO2). Вперше цю реакцію відкрив російський вчений М.І.Коновалов в 1888 р. (з тих пір вона названа його ім'ям). Алкани взаємодіють з розведеною азотною кислотою при нагріванні, утворюючи нітропохідні алканов: Сульфування. Димляча сірчана кислота (містить розчинений в ній SO3) з вищими алканами дає сульфокислоти.
2.2 Реакції розщеплення
Реакції розщеплення протікають при нагріванні (в присутності каталізаторів або без них).
1 Відщеплення водню (дегідрування). При нагріванні алканів у присутності каталізатора (СrО3) відбувається відщеплення атомів водню з утворенням ненасичених вуглеводнів.
2 Термічне розкладання (розрив зв'язків С-С і С-Н). Відомо, що алкани стійкі тільки при порівняно невисоких температурах. При нагріванні алканів до 500°С і вище, (без каталізаторів або в їх присутності) вони розкладаються з розривом зв'язків С-С і С-Н. В результаті відбувається утворення більш простих вуглеводнів – насичених і ненасичених. Цей процес називають крекінгом.
3 Ізомеризація. При цій реакції нерозгалужений вуглецевий ланцюг перетворюється на розгалужений. Це супроводжується розривом зв'язків С-С: Процес ізомеризації проходить при нагріванні в присутності каталізатора (А1С13). В цю реакцію вступають лише ті алкани, які в вуглецевому ланцюгу містять не менше чотирьох вуглецевих атомів.
2.3 Реакції окислення
При звичайних умовах алкани стійкі до дії навіть сильних окисників (КМnO4 , К 2Сr4 та ін.) Тому при додаванні до алканів водного розчину перманганату калію забарвлення розчину не змінюється. Однак при каталітичному окисленні (у присутності солей марганцю) і одночасному нагріванні відбувається окислення алканів (особливо вищих) з утворенням багатьох кисневмісних речовин (спиртів, кетонів, карбонових кислот та ін.)
На повітрі алкани горять з утворенням оксиду вуглецю (IV) і води. При цьому виділяється значна кількість теплоти. [5].
2.4 Нові напрямки у вивченні реакцій алканів
Останнім часом були знайдені нові гомогенні реакції алканів, що протікають у розчині або газовій фазі. Так, були відкриті реакції насичених вуглеводнів з електрофілами в розчинах сильних кислот або суперкислот [6, 7]. Як електрофільна частинка в розчинах кислот FSO3H-SbF5 або HF-SbF5 виступає протон, при атаці якого на алкан утворюється алконіевий іон СnН2n+3+, розпадається далі з розщепленням зв'язку С-H або С-С.
Електрофільне заміщення водню в алканах на нітрогрупу відбувається під дією NO2+ PF6- в діхлорметані при кімнатній температурі, а реакція алкана з Сl2 у SbF5-SO2C1F протікає навіть при -78 °.
Розчини флуораніла в HF-SbF5 окислюють метан, пропан, н-пентан [8]. При розчиненні Н2О2 або О3, в FSO3H—SbF5—SO2C1F виникають відповідно частки Н3О2+ і НО3+, які електрофільно атакують молекули алканів, в результаті чого утворюються різні кисневмісні похідні.
3. Металокомплексний каталіз
У 1969 р. відкрита перша реакція алкана з металокомплексами [9], в якій утворюються проміжні металоорганічні сполуки, тобто похідні, що містять зв'язок метал-вуглець [10]. Ця реакція відповідальна за H-D-обмін в метані, етані та вищих алканах в системі СnН2n +2—D2O—PtCl42-. Трохи пізніше була виявлена [11] здатність алканів окислюватися в системі з проміжним утворенням металоорганічних σ-комплексів платини. Періана та ін. [12-16] в своїх роботах повідомляли, що катіони Pd2+ в 96% сірчаної кислоти будуть каталізувати пряме окислення метану при 453 К до оцтової кислоти з утворенням бісульфату метану (попередника метанолу) і двоокису вуглецю, як побічних продуктів реакції відновлення.
Розуміння елементарних процесів, які входять в активацію і окислення метану, каталізуються катіонами Платини і Золота в сірчаній кислоті, було отримано за допомогою квантово-хімічних досліджень. Циглер і співавтори [17] вивчили механізм активації і функціоналізації метану за допомогою катіонів Pt2+ у сірчаній кислоти і визначили, що активація метану відбувається переважно через окисне приєднання.
Годдард і співавтори [15] вивчили детальніше систему і визначали вплив різних лігандів на стабільність і поведінку каталізатора. Вони показали, що активація зв'язку С-Н може відбуватися через електрофільне заміщення або окисне приєднання, залежно від лігандів на платинових центрах. Джонс та ін. [18] отримали експериментальні та теоретичні дані для перетворень метану в метанол сумішшю селенової кислоти і металевим золотом в 96%-ної сірчаної кислоти. Їх досліди показують, що метан піддається реакції електрофільного заміщення.
В роботах [19 - 23] представлений огляд стану досліджень в області каталізу біологічного окислення насичених вуглеводнів молекулярним киснем та моделювання цих процесів на основі комплексів металів.
Проблеми каталітичного окислення алканів розглядають у своїх роботах і українські автори. Провідне місце в Україні у сфері вивчення застосування металокомплексів для окислення алканів займає ІнФОВ НАН України, деякі результати цих досліджень представлені в [21].
Одним з перспективних напрямків металокомплексного каталізу є реакції за участю катіонів паладію. Можливі два механізми активації алканів комплексами паладію. Обидва починаються з утворення комплексу алкана з паладієм. Перший механізм є окисне приєднання метану по зв'язку С-Н, а другий – відрив атома водню координованим бісульфатним лігандом з одночасним формуванням зв'язку Pd-C

Рисунок 1 Приклад реакції окислення

Рисунок 2 Приклад реакції електрофільного заміщення
Металокомплексний каталіз базується на взаємодії субстрату з реагентом в координаційній сфері комплексу металу. У ряді випадків механізм аналогічний дії ферментів, що містять атом металу як кофактора. Металокомплексний каталіз може здійснюватися під дією гомогенних і гетерогенних металокомплексних каталізаторів. Перші присутні в розчині разом з реагентами і продуктами, другі здійснюють каталіз на поверхні, якщо вони не розчиняються в даному середовищі або різними способами нанесені на носій (полімер або неорганічний матеріал). Такі гетерогенізовані металокомплексні каталізатори мають високу селективність, відрізняються однорідністю активних центрів і легкістю їх модифікування, а також термостабильністю, тривалим строком служби та регенерованого. Виділяють такі особливості комплексів перехідних металів, визначають їх каталітичну активність:
1. Здатність утворювати комплекси з молекулами різних типів, які, входячи в координаційну сферу металу – комплексоутворювача, активуються, що забезпечує легкість їх подальшої взаємодії.
2. Утвореня комплексів з координуючим іоном або атомом металу знижує енергію зв'язку реагуючих молекул субстратів, що зменшує енергії активації їх подальших реакцій порівняно з некоординованими молекулами.
3. У координаційній сфері металу молекули змінюють свої кислотні або основні властивості і виникає можливість кислотно-основної взаємодії при тих значеннях рН, при яких вільна молекула не реагує.
4. Якщо є заборона по симетрії молекулярних орбіталей, що перешкоджає взаємодії молекул, то при реакції в координаційній сфері металу він може зніматися або значно послаблюватися.
5. У внутрішній сфері процеси, що протікають через перехідний стан, можуть бути дозволені і можуть перебігати з малими енергіями активації.
6. Металоорганічний каталіз дозволяє здійснити реакції багатоелектронного окислення і відновлення, в яких молекула субстрату в координаційній сфері відразу приймає або віддає кілька електронів. При цьому полегшуються процеси, в яких послідовний перенос електронів ускладнений через термодинамічні труднощі одно- або двоелектронної стадій. [24]
4. Характеристика неемпіричних методів квантової хімії
У роки зародження квантової хімії використовувалися напівемпіричні методи. Рух електронів в хімічних системах розглядалося тільки при фіксованому положенні ядер (адіабатичне наближення). Вивчалися молекули найлегших елементів - водню, гелію. Рішення рівняння Шредінгера навіть у цьому випадку пов'язане з трудомісткими розрахунками. [25]
В сучасних хімічних дослідженнях, завдяки поширенню потужних ЕОМ, широко застосовуються неемпіричні квантові розрахунки, в яких з експериментальних даних використовуються тільки заряди ядер атомів.
Неемпіричні методи слугують для вирішення найрізноманітніших завдань. Визначення рівноважної конфігурації ядер молекули вимагає пошук мінімуму на поверхні потенційної енергії, який проводять по точках, тобто багаторазово вирішують електронну задачу для різних конфігурацій ядер.
У перекладі з латинської ab initio означає з перших принципів
. Дійсно, до цієї групи належать методи, відповідно до
яких обчислення проводяться виключно на теоретичній базі, тобто без введення в розрахункову схему будь-яких параметрів,
отриманих експериментальним шляхом. При розрахунку всі величини мають конкретний фізичний зміст. Такими методами є: метод
Хартрі-Фока-Рутаана, різноманітні варіації конфігураційної взаємодії, методи теорії збудження, а також метод об'єднаних
кластерів. До переваг даного підходу слід віднести прийнятну точність розрахунку, відносну універсальність. Недоліком же
є ресурсомісткість процедури, тому група ab initio методів стала застосовуватися хіміками пізніше методів напівемпіричних
(Точніше, вони знаходили застосування, але лише до найпростіших систем,які не набагато перевершують по складності молекулу водню) [26]
Неемпіричні методи, в принципі, дають квантово-механічний опис станів системи, виходячи тільки з заданого числа електронів в ній, числа і зарядів ядер. Як правило, розрахунки молекул в рамках неемпіричних методів виконуються в адіабатичному наближенні, тобто роздільно вирішується так звана електронна завдання - визначення енергій і хвильових функцій електронів (електронних станів) при фіксованих положеннях ядер в просторі, і ядерна задача - розрахунок енергій і хвильових функцій ядер в полі, створюваному електронами в даному стані [27].
В даний час неемпіричні методи дозволяють вивчати особливості будови і взаємного розташування ППЕ, механізм реакцій, взаємодії молекули з електромагнітним випромінюванням, постійними або змінними зовнішніми впливами. Найбільш поширене застосування неемпіричних методів для малих молекул, для яких ці методи дозволяють часом отримати практично повний опис з точністю, близькою або навіть перевершує експериментальну. Розвиток обчислювальної техніки дозволяє проводити неемпіричні дослідження все більш складних систем, наприклад амінокислот або їх комплексів. Однак для послідовного аналізу виділеного класу споріднених сполук доцільніше використовувати методи, що враховують їх специфіку. Зазвичай молекули великого розміру описують за допомогою напівемпіричних методів квантової хімії або інших методів моделювання хімічних систем [27].
Неемпіричні (ab initio) методи, залишаючись найбільш точними і послідовними розрахунковими методами квантової хімії, тим не менше мають принципові недоліки, що ускладнюють їх широке застосування:
- велика трудомісткість розрахунків, пов'язана з тим, що час розрахунку росте як n4 (де n – число базисних атомних орбіталей (АО)), вимагає застосування суперкомп'ютерів вже для молекул з n ~ 200-250, що відповідає числу атомів N = 10-20 при використанні розширеного базису;
- доcі не вирішена проблема повного врахування кореляційної енергії Ecorr, величина якої в деяких випадках можна порівняти з енергією досить міцної ковалентного зв'язку, хоча і мала в порівнянні з повною енергією молекули (Etot) [28].
Головним інструментом дослідження у квантово-хімічному моделюванні є комп'ютер, на якому встановлена одна з програм для розрахунків за методом молекулярних орбіталей. До теперішнього моменту доступно кілька таких програм, які мають свої переваги і недоліки. Однією з таких програм є GAMESS (General Atomic and Molecular Electronic Structure System - система загального призначення для розрахунків атомних і молекулярних структур). Це некомерційне програмне забезпечення, яке його творці поширюють у вигляді бінарних кодів за умови регістрації та заповнення спеціальної анкети. Основною перевагою GAMESS-PC є висока швидкість роботи в порівнянні з іншими квантово-хімічними програмами, що важливо при дослідженнях складних молекулярних систем.
Деякі з можливостей програми GAMESS представлені нижче:
- Можливість розрахунку молекулярних хвильових функцій методом самоузгодженого поля в наближенні RHF, UHF, ROHF, GVB і MCSCF;
- Облік енергії електронної кореляції на основі теорії збурень, конфігураційного взаємодії, пов'язаних кластерів та функціоналу щільності;
- Можливість виконання напівемпіричних розрахунків методами MNDO, AM1 і PM3;
- Автоматична оптимізація геометрії, пошук перехідних станів з використанням аналітичних градієнтів;
- Рішення коливальної задачі - розрахунок частот валентних коливань ІЧ- та спектрів комбінаційного розсіювання;
- Обчислення молекулярних властивостей, таких як дипольний момент, електростатичний потенціал, електронна та спінова густина, аналіз заселеності по Маллікену і Левдіну;
- Можливість моделювання впливу розчинника.
Робота у програмі GAMESS полягає у складанні вихідного файлу з зазначенням всіх необхідних параметрів. Після створення файлу вихідних даних виклик програми проводиться з командного рядка, зручно використовувати Total Commander. Перегляд результатів відбувався за допомогою програми Chemcraft також за допомогою цієї програми можна задати приблизну геометрію молекули. Вигляд вікна програми Chemcraft показан на рисунку 3.
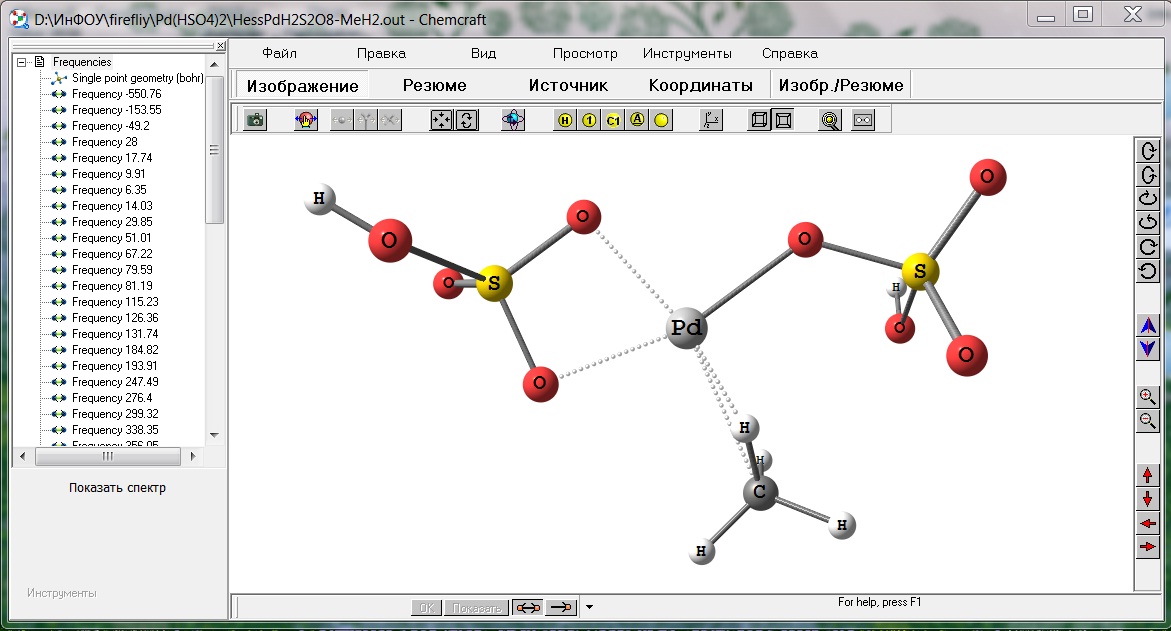
Рисунок 3 Інтерфейс Chemcraft
Висновки
Хоча сполуки перехідних металів відрізняються досить складною структурою і дорожнечею, в їх використанні перш за все привертає увагу висока селективність спільно з високою активністю каталізаторів. Саме тому дослідження властивостей перехідних металів і механізмів їх реакції є актуальними науковими задачами, розгляду яких і присвячена дана магістерська робота.
Таким чином, в магістерській роботі вирішені наступні завдання:
- Проведено дослідження реакцій активації метану, яка каталізується катіонами паладію (II), оточеного різними лігандами.
- Розраховані структури вихідних комплексів паладію та їх метанових похідних.
- Знайдено структури перехідних станів активації метану по двом можливим механізмам - окисне приєднання і відрив протона координованим бісульфатним лігандом з одночасним утворенням зв'язку Pd-C.
- Виконано оцінку бар'єрів активації досліджених реакцій, що відповідають двом можливим механізмам.
Подальша робота направлена на дослідження процесів активації як паладієвим комплексами з різними лігандами, так і іншими металокомплексами.
Перелік посилань
- Синев М. Ю., Корчак В. Н., Крылов О.В. Механизм парциального окисления метана: – Успехи химии, 1987, №5,сс.754 -792
- Шилов А.Е., Шульпин Г.Б. Активация и каталитические реакции алканов в растворах комплексов металлов: – Успехи химии, 1989, №1,сс.38 -57
- Алканы [Электронный ресурс] – Режим доступа: http://chemister.ru/Database/words-description.php?dbid=1&id=92
- Метан, Methane [Электронный ресурс] – Режим доступа: http://www.niikm.ru/articles/element_articles/methane/
- Предельные, или насыщенные, углеводоробы ряда метана(алканы или парафины) [Электронный ресурс] – Режим доступа: http://himiy.ucoz.ru/index/0-9
- Shilov A. E. Activation of Saturated Hydrocarbons by Transition Metal Complexes. Dordrecht: D. Reidel, 1984.
- Olah G. A., Parker D. G., Yoneda N. Angew. Chem., Int. Ed., 1978, v. 17, p. 909.
- Bedioni F., Farbre P.-L, Devynek J. Chem. Communs, 1982, p. 290.
- Гольдшлегер Н.Ф., Тябин М.Б., Шилов А.Е., Штейнман А.А. Журн. физ. химии, 1969, т. 43, с. 2174.
- Губин С. П., Шульпин Г. Б. Химия комплексов со связями металл — углерод. Новосибирск: Наука, 1984.
- . Гольдшлегер Н. Ф., Еськова В. В., Шилов А. Е., Штейнман А. А. Журн. физ. химии, 1972, т. 46, с. 1353.
- Periana, R. A.; Mironov, O.; Taube, D.; Bhalla, G.; Jones, C. J. Science 2003, 301, 814.
- Kua, J.; Xu, X.; Periana, R. A.; Goddard, W. A. Organometallics 2002, 21, 511.
- Xu, X.; Kua, J.; Periana, R. A.; Goddard, W. A. Organometallics 2003, 22, 2057.
- Jones, C. J.; Taube, D.; Ziatdinov, V. R.; Periana, R. A.; Nielsen, R. J.; Oxgaard, J.; Goddard, W. A. Angew. Chem., Int. Ed. 2004, 43, 4626.
- Zerella, M.; Kahros, A.; Bell A. T. J. Catal. 2005, in press.
- Zerella, M.; Mukhopadhyay, S.; Bell, A. T. Chem. Commun. 2004, 1948
- Gilbert, T. M.; Hristov, I.; Ziegler, T. Organometallics 2001, 20, 1183.
- Hristov, I. H.; Ziegler, T. Organometallics 2003, 22, 1668.
- Treutler, O.; Ahlrichs, R. J. Chem. Phys. 1995, 102, 346.
- Rudakov, E. S.; Yaroshenko, A. P.; Rudakova, R. I.; Zamashchikov, V. V. Ukr. Khim. Zh. 1984, 50, 680.
- Мацура В.А., Украинцев В.Б., Потехин В.В. Окисление молекулярным кислородом и гидрирование бензилового спирта в присутствии коллоидного палладия in situ// Журн. общей химии. 2000. Т.70. Вып. 12. С. 2058.
- Рядинская Н.Ю., Потехин В.М., Потехин В.В. Реакции третичных спиртов с тетрааквакомплексом палладия(II). Образование π-аллильных комплексов палладия // Журн. общей химии. 2001. Т.71. Вып. 8. С. 1242-1248.
- Металлокомплексный катализ [Электронный ресурс] – Режим доступа: http://www.chem.isu.ru/leos/base/eos15.html
- Квантовая химия на ПК: Компьютерное моделирование молекулярных систем : учеб.-метод. пособие / В. Б. Кобычев. – Иркутск : Иркут. гос. ун-т, 2006. – 87 с.
- Овчинников, М.Ю. История квантовой химии [Электронный ресурс] – Режим доступа: http://www.qchem.ru/materials/history/omyu_qchem/
- Химическая энциклопедия / «Советская энциклопедия», М. – 1988
- Блатов, В.А. Полуэмпирические расчетные методы квантовой химии/ В.А. Блатов A.П. Шевченко, Е.В. Пересыпкина/ «Универс-групп», 2005