
Реферат за темою випускної роботи
- Вступ
- 1. Способи переробки головної фракції сирого бензолу
- 2. Фізико–хімічні основи процесу переробки ГФСБ аміачним способом
- 3. Експериментальна частина
- 3.1 Методика виділення СS2 з ГФСБ.
- 3.2 Методика аналізу продуктів водної та органічної фаз
- 3.3 Розділення продуктів органічної фази – бензолу і дициклопентадієну
- 3.3.1 Проста перегонка
- 3.3.2 Перегонка з водяною парою
- 3.3.3 Перегонка під вакуумом
- 4. Аналіз експериментальних даних
- 4.1 Процес виділення СS2 з ГФСБ
- 4.2 Процес розділення продуктів органічної фази – бензолу і дициклопентадієну
- 4.2.1 Проста перегонка
- 4.2.2 Перегонка з водяною парою і під вакуумом
- 5. Пілотна установка для розділення продуктів органічної фази
- Висновки
- Перелік посилань
Вступ
Сирий бензол, що отримують в процесі коксування кам’яного вугілля, у подальшому переробляється з метою отримання чистих продуктів. Схема переробки має стадію попередньої ректифікації, що забезпечує роз’єднання низькокиплячих та висококиплячих сірчистих та ненасичених сполук. Суміш низькокиплячих компонентів, що отримується при цьому, зветься головною (ГФСБ), або сірковуглецевою фракцією. Вихід головної фракції складає 2,0-3,5%. На підприємствах, що переробляють бензол, який отримано з сірчистого донецького вугілля, головна фракція вміщує 20-35 % сірковуглецю, 15-35 % циклопентадієну, 20-50 % бензолу, 2-5 % дициклопентадієну, до 10 % ненасичених (олефіни C5H10)и до 4,0 % насичених сполук (циклопентан й гомологи, пентан, гептан) [1].
Потенційні ресурси ГФСБ в Україні складають 10 тис. т на рік. Кількість сірковуглецю, що вміщується у фракції – 3 тис. т на рік.
Проте, на даний час, не існує простого, надійного та безпечного способу переробки ГФСБ. [2]. Отже, нагальною є потреба розробки нового способу переробки цієї фракції.
У даній роботі описано спосіб переробки ГФСБ з використанням хімічного реагенту аміаку, наведено аналіз експериментальних даних по розділенню отримуваних продуктів.
Мета: розділення продуктів органічної фази, отриманої після обробки ГФСБ аміаком.
Актуальність:
- Необхідність розробки способу розділення речовин органічної фази первинної переробки ГФСБ;
- Високий попит на чисті вуглеводні сполуки (бензол та дициклопентадієн) для промислового синтезу.
Об’єкт дослідження: органічна фаза – суміш бензолу та дициклопентадієну, - отримана при очищенні ГФСБ хімічним способом.
1. Способи переробки головної фракції сирого бензолу
Склад ГФСБ залежить від умов її відбору, а також від складу вихідної сировини.
ГФСБ, що виділяється при ректифікації сирого бензолу, характеризується наступними показниками [1]:
Густина при 20 °С, кг/м3 – 930-980
Розгонка, °С:
початок кипіння – 30-38
температура при відгонці 95% – 66-75
У відповідності до зміни складу головної фракції змінюється її вихід. При отриманні концентрованої головної фракції, вихід її зазвичай не перевищує 2-3 %. За високої температури наверху колони, отримують головну фракцію розбавлену бензолом, та вихід її досягає 4-6 %.
Переробка головної фракції - складна технологічна операція, що пов’язана зі значними втратами та труднощами при витягуванні бензолу. Тому надзвичайно важливо прагнути до отримання якнайбільш концентрованої фракції [2].
Чим вища температура унизу колони, тим більш повно проходить витягування сірковуглецю. Підвищити температуру хоча б до 90 °С можливо лише при великому рефлексному числі.
Головна фракція при зберіганні набуває яскраво виражений кислий характер, причому зі збільшенням тривалості зберігання кислотність її зростає. Встановлено, що зростання кислотності обумовлено утворенням перекисів, та для стабілізації ГФСБ при зберіганні, її необхідно інгібітувати.
До головної фракції переходить 41-46 % циклопентадієну, до того ж мають місце прямі технологічні втрати. Залишкова кількість циклопентадієну у фракції БТКС складає зазвичай 0,1-0,15 %.
Близькість температур кипіння сірковуглецю та циклопентадієну (42,5 та 46,5 ºС відповідно) при приблизно однаковому їх вмісту дуже ускладнює виділення концентрованої головної фракції тільки ректифікацією.
Промислове застосування знайшов спосіб переробки головної фракції методом термічної полімеризації, що заснований на властивості циклопентадієну утворювати під впливом нагріву дициклопентацієн з різко відмінною від інших компонентів фракції температурою кипіння 166,6ºС. Самодовільний перехід циклопентадієну в дициклопентацієн призводит ь до того, що з перебігом часу склад фракції та характеристика її розділення значно змінюються [1,2].
Вперше в промисловості процес термічної полімеризації сірковуглецевої фракції був здійснений цехом ректифікації Макіївського коксохімічного заводу.[ [1]. Процес термічної полімеризації здійснюється в агрегаті періодичної дії при температурі, що не перевищує 120°С при повному поверненні конденсату парів, що утворюються. Головна фракція завантажується до кубу ректифікації, в якому, завдяки підігріву глухою парою, здійснюється полімеризація циклопентадієна в димер, а також його сополімеризація з деякими ненасиченими сполуками. Продукти, що не полімеризуються та не виділяються, конденсуються та повертаються до кубу.
Тривалість процесу термічної полімеризації головної фракції залежить переважно від складу фракції і становить 12 - 24 години. При проведенні полімеризації безперервно змінюється склад фракції внаслідок утворення полімерів, а також втрати легкокиплячих вуглеводнів крізь повітряники. Сприятливо процес полімеризації проводити за можливістю швидко, щоб мінімізувати неминучі втрати сірковуглецю крізь повітряники. Закінчення процесу полімеризації визначається за припиненням зміни складу завантаження кубу [1,3-5].
Спеціалістами Вінницького національного технічного університету запропонований спосіб витягнення сірковуглецю із головної фракції шляхом обробки її водним розчином амінів або амідів з отриманням відповідних дітіокарбаматів. Даний спосіб реалізується у дві стадії. На першому ступені утворюється сіль диалкілдитіокарбаминової кислоти за реакцією:
 (1.1)
(1.1)
Alk – CH3, C2H5, C3H7, C4H9; M – K, Na, NH4
На другому ступені сіль перетворюється у відповідні дитіокарбамати деяких 3-d металів за наступною схемою:
 (1.2)
(1.2)
М ' – Cu2+, Co2+, Ni2+, Zn2+, Mn2+, Fe2+, Sn2+, Pb2+; An – Cl-, NO-, 1/2 SO42- [7].
2. Фізико–хімічні основи та хімізм процесу переробки ГФСБ аміачним способом
Як показує літературний огляд (пункт 1), на наш час існує мало способів переробки ГФСБ, а ті способи, що маються, далекі від досконалості в технологічному, екологічному та економічному аспектах.
На наш час спосіб термічної полімеризації не застосовується, внаслідок його пожежо- і вибухонебезпечності, не екологічності, великих (до 30%) втрат основних компонентів та значних економічних витрат [1,2].
Спосіб, розроблений спеціалістами Вінницького національного технічного університету, ще не вийшов за рамки лабораторних випробувань та не був застосований в технологічних умовах. Окрім того, аміни є відносно дорогими реагентами і в Україні не виробляються.
Дослідження були спрямовані на вивчення способу витягування сірковуглецю з головної фракції шляхом зв’язування його хімічним реагентом з отриманням цінних хімічних продуктів, безпечних при застосуванні, збереженні та транспортировці [4].
У якості реагенту для витягування сірковуглецю з головної фракції був обраний водний розчин аміаку. Вибір аміаку обумовлений тим, що він є відносно недорогим та доступним. Окрім того, аміак може вибірково взаємодіяти з сірковуглецем головної фракції. В той же час, сполуки, що одержуєть при взаємодії сірковуглецю з аміаком, можуть бути використані для виробництва важливих і цінних хімічних продуктів.
Взаємодія відбувається за такими схемами:
Схема 1, t ≥ 120 °С, утворюється тіомочевина
CS2 + 2NH3 → H2N−CS−NH2 + H2S (2.1)
Схема 2, t = 113 °С, утворюється роданід амонію
CS2 + 2NH3 → NH4−S−C≡N + H2S (2.2)
Схема 3. Ізомеризація роданіду амонію в тіомочевину, t ≥ 160
NH4−S−C≡N → H2N−CS−NH2 (2.3)
Схема 4. Утворення сірчистого амонію
H2S + 2NH3 → (NH4)2S (2.4)
За температури 20-25 ° С взаємодія сірковуглецю і аміаку протікає надзвичайно повільно, а технологічно вигідна реакція йде при підвищеній температурі та тиску. У той же час, збільшення температури неприпустимо, оскільки компоненти реакційної суміші – сірковуглець і аміак – легколетючі й отруйні компоненти. Дотримуючись цих вимог, процес проводили в герметичному автоклаві.
Для визначення оптимальних умов проведення процесу взаємодії ГФСБ та аміаку був досліджений вплив часу контакту на ступінь витягування CS2 . Після 3 годин спостерігається незначна зміна ступеня витягування сірковуглецю, тому оптимальним часом для проведення подальших дослідів був прийнятий час – 3 години.[6].
Температура кипіння сірковуглецю дорівнює – 42,5 °С а водний розчин аміаку має високий парціальний тиск. Очевидно, що взаємодія між CS2 и NH3 в інтервалі температур 80-150°С протікає в газовій фазі. Тиск, що створюється в автоклаві, сприятливо впливає на реакції 2.1-2.3, оскільки вони йдуть в газовому середовищі зі зменшенням числа молей компонентів системи і супроводжуються поглинанням проміжних продуктів (дітіокарбамінат амонію, тіомочевина, роданід амонію, сірчистий амоній), що утворюються, водною фазою. За результатами дослідів видно, що при зміні співвідношення NH3: CS2 с 2:1 до 7:1 ступінь витягування сірковуглецю збільшується від 77,3 % до 99,8 %. При стехіометричному співвідношенні NH3: CS2 = 2:1 ступінь витягування CS2 складає 77,3%.
Аналіз отриманих даних показав, що при збільшенні температури з 85 до 150 °С ступінь витягування зростає з 75,5 до 98 % (при співвідношенні NH3: CS2= 5:1). При температурі 115 °С досягається практично повне витягування сірковуглецю з органічної фази до водної фази в формі роданистих сполук (при співвідношенні NH3: CS2= 7:1) [6].
Таким чином, було обрано мольне співвідношення NH3: CS2= 7:1, час контакту 3 години, температура 120 °С.
3. Експериментальна частина
В лабораторних умовах провели експериментальні дослідження, в яких використовували:
- головну фракцію сирого бензолу складу, %: CS2 – 32,9;
циклопентадієн – 0,9; дициклопентадієн – 33,1;
бензол – 25,6;
легкі компоненти (ненасичені, алкани, циклоалкани) – 7,5. - водний розчин 25%-вого аміаку.
3.1 Методика виділення CS2 з ГФСБ
Як було описано вище, для проведення реакції використовували лабораторну установку &ndash автоклав. Автоклав складається з корпусу з кришкою, в який поміщається стакан з реакційною сумішшю.
У склянку поміщали розрахункову кількість ГФСБ і 25%-вого аміаку. Потім склянку опускали в автоклав і щільно закривали кришкою. Автоклав ставили в термостат, нагрітий до певної температури. Далі автоклав нагрівали і витримували певний час.
Після витримки автоклав діставали на повітря, потім охолоджували проточною водою 30 хвилин. Потім суміш переливали в ділильну воронку і розділяли на дві фази: водну і органічну [9].
3.2 Методика аналізів продуктів водної та органічної фаз
Аналіз аміачно – сольового розчину
Для аналізу системи сірчистий амоній – роданистий амоній – тіомочевина була розроблена методика, сутність якої полягає в роздільному визначенні сірчистого і роданистого амонію. Визначення роданіду амонію проводять титруванням азотнокислою ртуттю (II) з попереднім осадженням сірчистих сполук оцтовокислим кадмієм Cd(CH3COO)2.Визначення сульфідів і тіомочевини складається з визначення загальної сірки шляхом титрування з I2, осадження сульфідів Cd (CH3COO)2 і визначення тіомочевини. Кількість сульфідів визначають за різницею об’єму, який пішов на титрування загальної сірки та тіомочевини.
Аналіз органічної фази
Аналіз органічної фази проводили хроматографічним способом на хроматографі КристаллЛюкс-4000.
Умови хроматографування [5]:
- Газ-носій – гелій;
- Фаза – ПЕГА, 15%;
- Носій – дінахром;
- Температура термоста – 65 °С;
- Швидкість газа – носія – 40 мл / хв;
- Довжина колонки – 4,5 м.
ККомпоненти головної фракції визначаються за хроматограмою (рис. 3.1). Основні компоненти суміші проявляються в такій послідовності:
- легкокиплячі(до С8)насичені вуглеводні;
- легкокиплячі (до С5)ненасичені вуглеводні;
- сірковуглець (час удержування відносно бензолу 0,21);
- циклопентадієн (час удержування відносно бензолу 0,25);
- н-нонан (час удержування відносно бензолу 0,72);
- бензол.

Рисунок 3.1–Хроматограма головної фракції сирого бензолу з введенням 12,3 % н-нонану
Дослід вважається закінченим після того, як пік бензолу повністю вималюється на хроматограмі.
В конічній колбі з притертою пробкою ємкістю 50 мл зважують з точністю до 0,0002 г 1 – 1,5 г н-нонана, після чого додають з точністю до 0,0002 г проби, що досліджується, в кількості 10–15 г. Після цього розраховується вміст н-нонана в отриманій суміші, який повинен складати 10 – 15 % (за масою). Пробу ретельно збовтують в конічній колбі та за допомогою мікрошприца вводять до хроматографа.
Розрахунок вмісту сірковуглецю: визначення вмісту сірковуглецю проводиться шляхом зіставлення площей піків н-нонана і сірковуглеця, які визначаються за хроматограмою.
РРозрахунок проводиться за наступною формулою [5] (3.1):
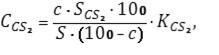
де СCS2 – концентрація сірковуглеця у головній фракції, % (за масою);
С – концентрація н-нонана в суміші, що аналізується, % (за масою);
SCS2 – площа піка сірковуглецю, мм2;
S – площа піка н-нонана, мм2;
KCS2 – калібровочний коефіцієнт
3.3 Розділення продуктів органічної фази – бензолу та дициклопентадієну
У даній роботі були розглянуті і проведені дослідження трьох способів розділення органічної фази: простої перегонки, перегонки з водяною парою і під вакуумом.
3.3.1 Проста перегонка
Органічна фаза після витягнення сірковуглецю представляє собою суміш бензолу, дициклопентацієну (ДЦПД), легких компонентів, циклопентадієну (ЦПД) та сірковуглецю, що не прореагував. Густина 0,880 г/см3.
Склад органічної фази представлений у таблиці 3.1
|
№ досліду |
t,°С |
Вміст, % |
||||
|
CS2 |
Бензол |
ЦПД |
ДЦПД |
лeгкі |
||
|
1 |
85 |
10,6 |
33,5 |
0,04 |
43,8 |
6,2 |
|
2 |
100 |
5,8 |
36,4 |
0,07 |
45,6 |
6,9 |
|
3 |
115 |
2,1 |
36,8 |
0,07 |
46,6 |
7,3 |
|
4 |
160 |
0 |
38,2 |
1,34 |
49,3 |
11,3 |
Таблица 3.1 – Склад органічної фази в залежності від температури проведення експериментів
Концентрація бензолу при відсутності втрат повинна збільшуватись пропорційно кількості витягненого сірковуглецю, тому що в умовах досліду бензол не бере участі у хімічних перетвореннях. Це підтверджують експериментальні значення для температур 85-115º С.
Проста перегонка здійснюється при атмосферному тиску, у звичайних перегінних приборах. Ведення процесу полягає у наступному: органична фаза в перегінній колбі нагрівається на пічці й переходить у пару, далі по відводній трубці та нисходячому холодильнику конденсується до приймача. В колбі залишається високолетючий залишок.
Подальший аналіз складу отриманих сумішей органічної фази, а саме: бензол у приймачі та дициклопентадієн, що залишився у колбі, проводиться хроматографічним способом на хроматографі КристалЛюкс-4000.
3.
3.2 Перегонка з водяною пароюПри нагріванні до високих температур продукти реакції можуть розкладатися.
Навідміну від простої перегонки, застосування гострої пари дозволяє провести розділення речовин за більш низької температури, внаслідок чого зменшуються втрати легколетючих компонентів.
Лабораторна установка для перегонки органічної фази, що отримана після взаємодії сірковуглецю ГФСБ з аміаком, зображена на рис. 3.2.

Рисунок 3.2 – Лабораторна установка для перегонки з водяною парою органічної фази, що отримана після взаємодії сірковуглецю ГФСБ з аміаком.
1 – тригорла колба, 2 – штатив, 3 – колба, 4 – піч, 5 – приймач конденсату, 6 – затискач,
7 – кипілки, 8 – пробки, 9, 11 – термометр, 10 – дефлегматор-«ялинка», 12 – холодильник,
13 – приймач бензолу, 14 – алонж, 15 – склянка Дресселя, 16 – переходная трубка
Головну фракцію, що отримана після взаємодії з аміаком, нагрівали на водяній бані у тригорлій колбі 1, що закріплена на штативі 2.
Перегонку вели з гострою парою, що отримували в колбі 3 й по перехідній трубці 16 направляли до тригорлої колби 1. Суміш парів води та бензолу, що утворилася, направляється крізь дефлегматор-«ялинка» 10, проходить холодильник 12, звідки, вже конденсована суміш, стікає по алонжу 14 до приймача 13. Також, крізь алонж 14 проходить пара Н2S, потрапляє до склянки Дресселя 15, де поглинається аміаком.
3.
3.3 Перегонка під вакуумомРечовини, що досліджуються,за атмосферного тиску переганяються з великими втратами, окрім того, ДЦПД при збільшенні температури може деполімерізуватися.
У той же час, застосування для розділення продуктів органичної фази водяної пари у промисловості буде пов'язано з утворенням великої кількості стічних вод. З метою усунення цих недоліків застосували перегонку під вакуумом.
На практиці перегонку під вакуумом проводять зазвичай при тиску від 50 до 1 мм рт. ст.
Апаратурне оформлення для перегонки під вакуумом подібне апаратурі для перегонки при атмосферному тиску, але з деякими додатковими деталями. Прибор для перегонки під вакуумом складається з наступних частин: печі для нагріву колби, перегінної колби, холодильника, приймача дистиляту, вакуум-насосу та приладдя для вимірювання й регулювання тиску.
Лабораторна установка для перегонки органічної фази під вакуумом наведена на рис. 3.3.

Рисунок 3.3 – Лабораторна установка для перегонки під вакуумом органічної фази, отриманої після взаємодії сірковуглецю ГФСБ з аміаком
1 – перегінна колба, 2 – штатив, 3 – піч,4 – пробки, 5,7 – термометр, 6 – дефлегматор-«ялинка», 8 – холодильник, 9 – алонж, 10 – приймач бензолу, 11 – склянка Дресселя
Відмінність ц процесі перегонки під вакуумом від перегонки з водяною парою полягає у тому, що для створення вакууму в першому випадку, до приймача під'єднують товстостінною резиновою трубкою манометр та насос.
4. Аналіз експериментальних даних
4.1 Процес виділення СS2 з ГФСБ
Внаслідок протікання реакції утворюється водна та органична фази наступного складу (рис. 4.1):

Рисунок 4.1 – Склад водної та органичної фаз.
4.2 Процес розділення продуктів органічної фази – бензолу і дициклопентадієну
4.2.1 Проста перегонка
В результате проведения простої перегонки був отриман відгон бензолу у приймачі й залишок в колбі.
Хроматографічний метод анализу дозволив визначити сосклад цих проб:
• у приймачі: 0,76% СS2, 3,97% ЦПД, 74,69% бензолу, 1,18% ДЦПД;
• залишок в колбі: 0,05% СS2, 0,03% ГПД, 48,31% бензолу, 38,05% ДЦПД.
Вихідна органічна фаза мала наступний склад:
2,10% СS, 0,07% ЦПД, 36.8% бензолу, 46,6% ДЦПД.
Дициклопентадієн здатен при підвищенні температуры до 115º С самодовільно переходити у мономір, отже необхідно вести перегонку за якнайменшої температури. Для зниження температури були використані інші методи - перегонка з водяної парою й під вакуумом.
4.2.2 Перегонка з водяною парою й під вакуумом
Хроматограми, отримані внаслідок аналізу продуктів перегонки головної фракції з водяною парою, представлені на рис. 4.2 – 4.4.

Рис. 4.2 – Хроматограма вихідної головної фракції, отриманої після обробки головної фракції аміаком
Згідно з результатами аналізу хроматограми, у вихідній пробі міститься 62,8% бензолу, 25,8% дициклопентадієну, 0,19% сірковуглецю.

Рис. 4.3 – Хроматограма проби у приймачі бензолу, що отримана посля перегонки ГФСБ з водяною парою
Згідно з даними аналізу, у приймачі міститься 81,7% бензолу, 3,7% дициклопентадієну, 0,59% сірковуглецю.

Рис. 4.4 – Хроматограма залишку в колбі після перегонки ГФСБ з водяною парою.
При аналізі хроматограми отримали наступні показники: кількість дициклопентадієну 55,7%, бензолу 30,7%, сірковуглецю 0,005%.
Результати позділення бензолу й дициклопентадієну різними методами надані у таблиці 1, %:
| Перегонка з водяною парою | Перегонка під вакуумом | ||
| Вихідна проба |
Бензол |
62,8 |
58,82 |
|
ДЦПД |
25,8 |
21,53 |
|
|
СS2 |
0,19 |
0,19 |
|
| У приймачі |
Бензол |
81,7 |
90,04 |
|
ДЦПД |
3,7 |
0,22 |
|
|
СS2 |
0,59 |
0,01 |
|
| Залишок в колбі |
Бензол |
30,7 |
17,68 |
|
ДЦПД |
55,7 |
42,00 |
|
|
СS2 |
0,005 |
2,97 |
|
Таблиця 1 - Результати перегонок з водяною парою й під вакуумом
Порівнюючи результати дослідних даних, можна зробити висновок, що перегонка з водяною парою й під вакуумом є досить ефективними, при їх застосуванні отримуємо 82-91%-вий бензол та насичену дициклопентадієном (42-56%) залишкову фракцію.
5. Пілотна установка для розділення продуктів органічної фази
Для розділення продуктів органічної фази пропонується застосовувати ректификаційну установку.
В ректифікаційних колонах вихідна суміш в результаті масообміну між паровою й рідкою фазами, що рухаються противотоком, розділяється на два компоненти: дистилят, який насичений більш летким компонентом, та кубовий залишок, що містить набагато менше леткого компоненту.
Розрахунок насадочної ректифікаційної колони безперервної дії.
Розрахунок ректифікаційної колони зводиться к визначенню геометричних розмірів - діаметру й висоти. Установка процює при атмосферному тиску та з доволі агресивними рідинами, тому приймаємо у якості насадки керамічні кільця Рашига розміром 10 × 10 × 1,5, питома поверхня насадки а = 440 м2/м3, вільний об'єм ε = 0,7 м3/ м3, насипна щільність ρ = 700 кг/м 3 [11].
На рис. 5.1 наведена принципова схема насадочної ректификаційної установки безперервної дії.

Рис. 5.1 – Принципова схема насадочної ректификаційної установки безперервної дії
1 – перегінний куб, 2 – насадочна ректификаційна колона, 3 – дефлегматор, 4 – розділювач потоку, 5 – теплообмінник, 6 – приймач дистиляту, 7 – ємність для кубової рідини
Вихідну суміш і водяну пару подають в перегінний куб 1, звідки пара надходить до ректифікаційної колони 2.
Стікаючи вниз колони, рідина взаємодіє з парою та піднімається наверх колони. Початковий склад пара приблизно дорівнює складу кубового залишку, тобто містить малу кількість легколеткого компоненту (бензолу). В результаті масообміну з рідиною, пара збагачується легколетким компонентом. Для більш повного збагачення, верхню частину колони зрошують флегмою (відповідно до заданого флегмовом числом). Частина конденсату виводиться з дефлегматора у вигляді готового продукту поділу – дистиляту, який охолоджується до теплообміннику 5 і надходить у приймач дистиляту 6.
З нижньої частини колони, а також з нижньої частини перегінного кубу, безперервно відводиться кубовий залишок – продукт, збагачений важколетким компонентом (дициклопентадієном). Кубовий залишок надходить до ємності для кубової рідини 7.
Розрахунок пілотної установки розділення продуктів органичної фази тут не наводиться, внаслідок його громіздкісті.
В результаті розрахунку: D = 6 см, Hнасадки = 1,5 м.
ВИСНОВКИ
- Виконано аналітичний огляд способів переробки ГФСБ, в результаті якого встановлено, що на даний момент існує мало способів переробки ГФСБ, а ті способи, що маються, далекі від досконалості в технологічному, екологічному та економічному аспектах. Метод термічної полімеризації, що раніше застосовувався у промисловості, зараз не використовується, внаслідок його пожежо– і вибухонебезпечності, неекологічності, великих (до 30%) втрат основних компонентів і значних економічних витрат.
- Розглянуто фізико–хімічні основи переробки ГФСБ аміачним способом. Даний хімічний метод дозволяє практично повністю (97-99%) виділити сірковуглець з ГФСБ (при співвідношенні СS 2:NH 3 = 1:7, часу контакту 3:00 і температурі 120 °С). Крім того, обраний спосіб є екологічно безпечним, оскільки на першій стадії відбувається зв'язування токсичного сірковуглецю.
- Показано, що для розділення продуктів можна застосовувати різні види перегонок. Однак з'ясовано, що з точки зору ефективності ці способи істотно відрізняються.
- При простій перегонці розділення відбувається при температурі 80 °С, але через нестійкість температур і великі втрати цінних хімічних компонентів, її застосування недоцільно.
- Перегонка з гострою парою показала наступні результати у вигляді чистих продуктів: 82%-вий бензол і 56%-вий ДЦПД. Недолік цього методу полягає в тому, що застосування водяної пари пов'язане з додатковими витратами на обладнання для виробництва пари, а отже з підвищенням собівартості одержуваних продуктів.
- Дослідження показали, що найбільш технологічним і економічним є спосіб перегонки під вакуумом (отримано відгін бензолу з концентрацією до 92% і ДЦПД - 42%). Надалі планується проведення серії дослідів перегонки при низькому тиску, тому що цей метод не потребує постійного виробництва додаткового технологічного агента (в даному випадку пари) і його застосування не пов'язано з утворенням стічних вод, що потребують додатковогго очищення.
При написанні даного реферату магістерська робота ще не закінчена. Остаточне завершення – грудень 2012 р. Повний текст роботи та матеріали за темою можуть бути отримані у автора або його керівника після зазначеної дати.
ПЕРЕЛІК ПОСИЛАНЬ
- Лейбович Р.Е., Обуховский Я.М., Сатановский С.Я. Технология коксохимического производства. – М.: Металлургия, 1966. – 463 с.
- Коляндр Л.Я. Новые способы переработки сырого бензола. – М.: Металлургия, 1976. – 192 с.
- Карлинский Л.Е. Выделение циклопентадиена из головной фракции сырого бензола / Л.Е. Карлинский, З.А. Бучкина, М.К. Горлов и др. // Кокс и химия. – 1968 – № 11. – С. 30 – 34.
- Химия и технология сероуглерода/ А.А. Пеликс, Б.С. Аранович, Е.А. Петров, Р.В. Котомкина. – Л.: Химия, 1986. – 224 с.
- Глузман Л.Д. Лабораторный контроль коксохимического производства/ Л.Д. Глузман. М.: Металлургия, 1968. – 473 с.
- О возможности извлечения сероуглерода из головной фракции сырого бензола химическим методом / И.Г. Крутько,А.В. Кипря, В.А. Колбаса, А.С. Комаров, К.А. Сацюк // Вісник Національного технічного університету «Харківський політехнічний інститут». Збірник наукових праць. Тематичний випуск «Хімія, хімічна технологія та екологія». – Харків: НТУ «ХПІ». – 2010. – № 11. – С. 43 – 47.
- Титов Т. С., Пелишенко С. В., Гордиенко О. А., Петрук Р. В.,Сандомирский О. В. Экологические вопросы коксохимических производств и пути их решения. Тези доповідей, 1 том, V Міжнародна науково-технічна конференції студентів, аспірантів та молодих вчених,Серія: хімія та сучасні технології, 2011. – 564 с.
- Крутько И.Г., Кіпря О.В., Комаров А.С. Извлечение сероуглерода из головной фракции сырого бензола раствором аммиака. Состав и свойства органической и водной фаз. Наукові праці Донецького національного технічного університету. Серія: Хімія і хімічна технологія, 2011. – Випуск 17 (187). – с. 167 – 171.
- Препаративная органическая химия. Под ред. Н. С. Вульфсона, М.: Химия, 1964 г., 908 с.
- Людмирская Г. С., Барсукова Т. А., Богомольный А. М. Равновесие жидкость – пар: Справ. изд. Под ред. А. М. Богомольного. Л.: Химия, 1987. – 336 с.
- Основные процессы и аппараты химической технологии: Пособие по проектированию/Г. С. Борисов, В. П. Брыков, Ю. И. Дытнерский и др. Под ред. Ю. И. Дытнерского, 2-е изд., перераб. и допол. М.: Химия, 1991. - 496 с.
- Курсовое проектирование по предмету «Процессы и аппараты химической промышленности»: Учеб. пособие для учащихся техникумов/ М.Н. Кувшинский, А. П. Соболева. – 2-е изд., перераб. и допол. – М, 1980. – 223 с.